We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Blood group antigen proteins Candidate cardiovascular disease genes Disease related genes FDA approved drug targets Plasma proteins Predicted secreted proteins
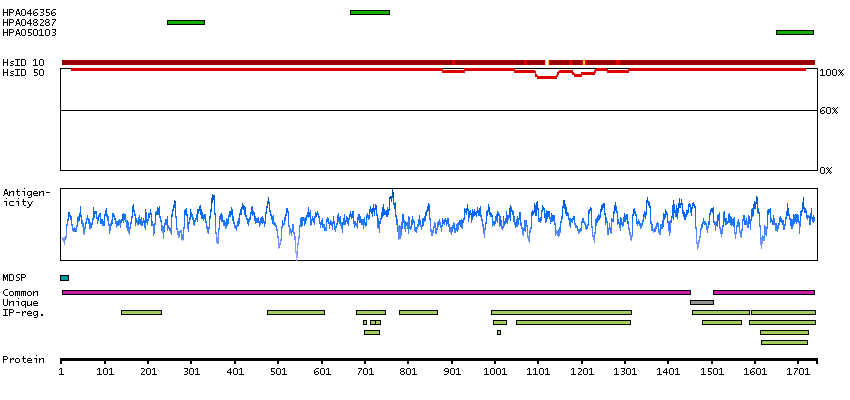
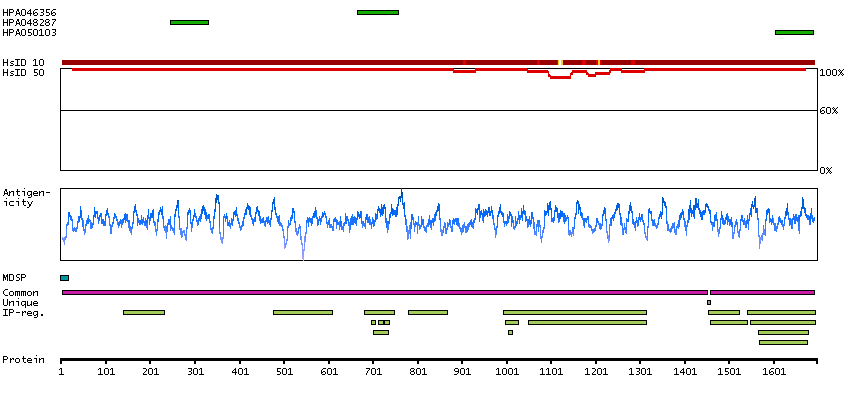
Cytoplasmic expression in liver and positivity in plasma in several tissues.
DATA RELIABILITY
Data reliability description
Caution, targets protein from more than one gene. Secreted protein, tissue location of RNA and protein might differ and correlation is complex. Presumed off target binding observed and disregarded.
This gene encodes the acidic form of complement factor 4, part of the classical activation pathway. The protein is expressed as a single chain precursor which is proteolytically cleaved into a trimer of alpha, beta, and gamma chains prior to secretion. The trimer provides a surface for interaction between the antigen-antibody complex and other complement components. The alpha chain is cleaved to release C4 anaphylatoxin, an antimicrobial peptide and a mediator of local inflammation. Deficiency of this protein is associated with systemic lupus erythematosus and type I diabetes mellitus. This gene localizes to the major histocompatibility complex (MHC) class III region on chromosome 6. Varying haplotypes of this gene cluster exist, such that individuals may have 1, 2, or 3 copies of this gene. Two transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Nov 2014]