We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
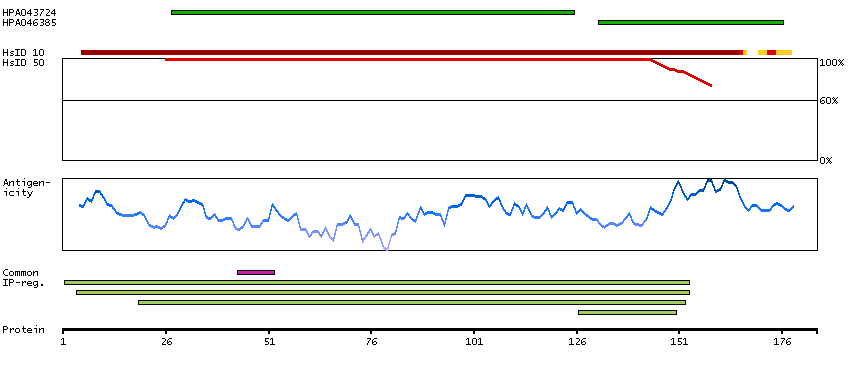
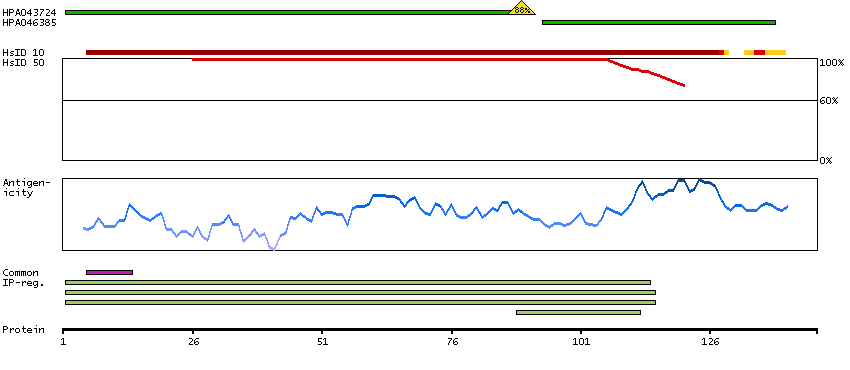
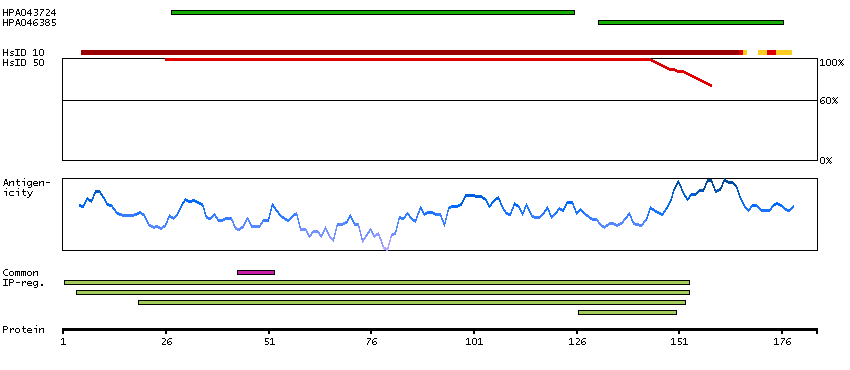
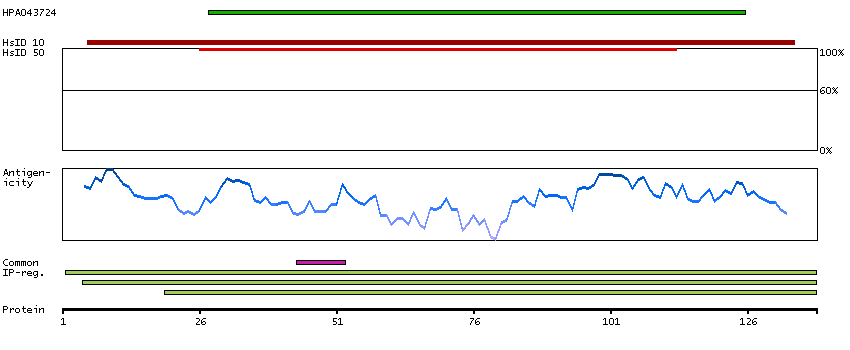
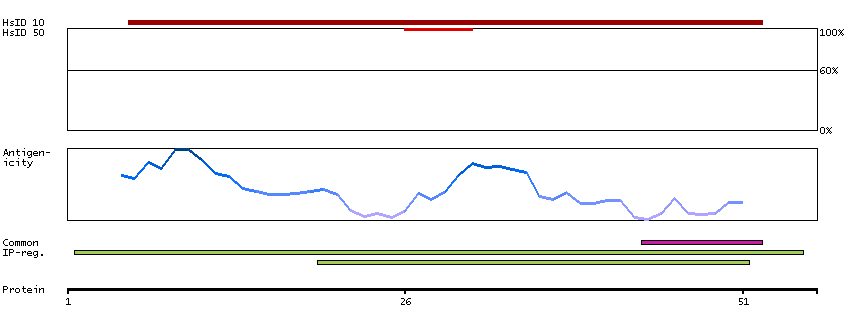
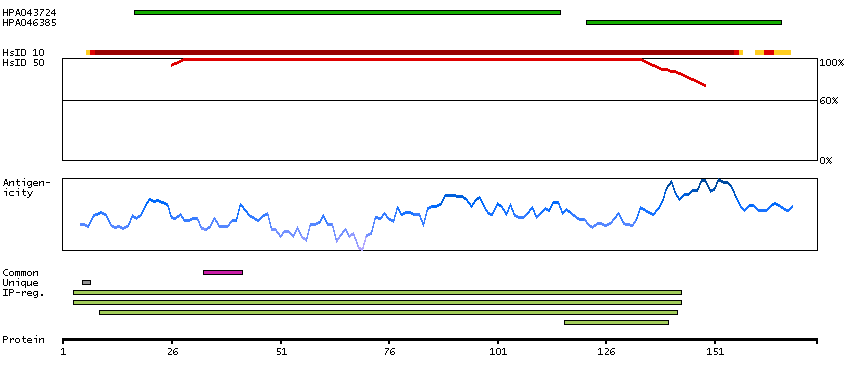
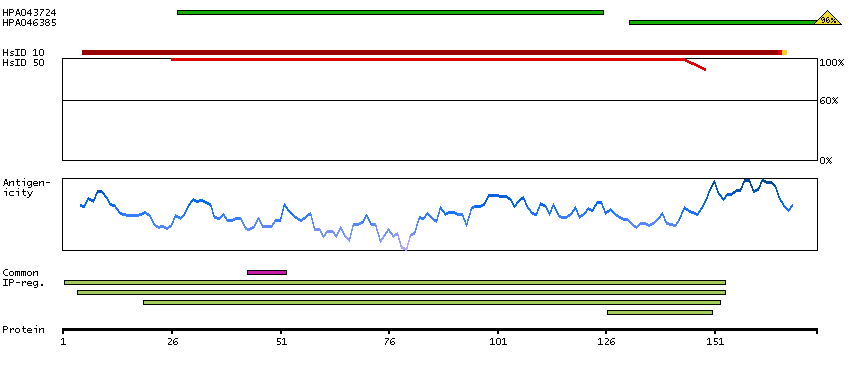
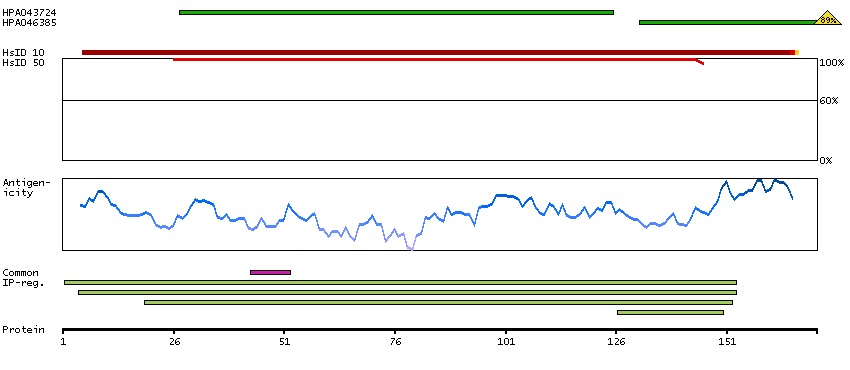
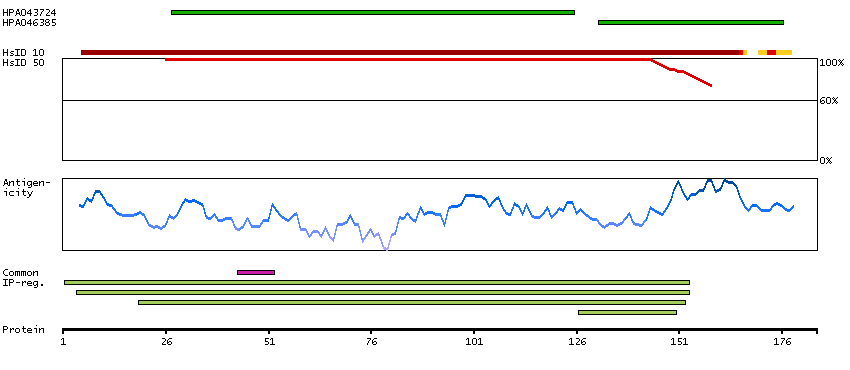
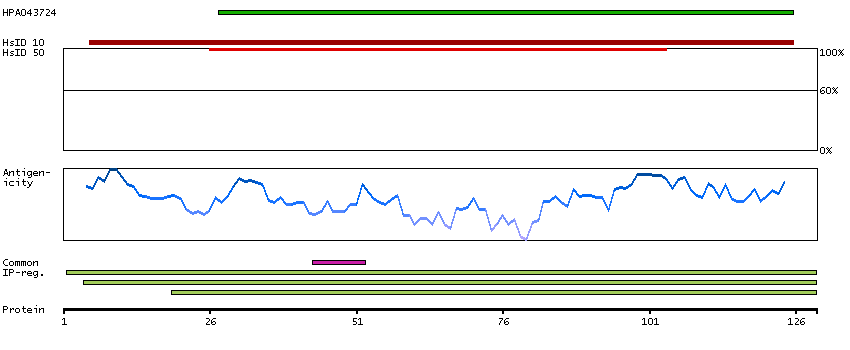
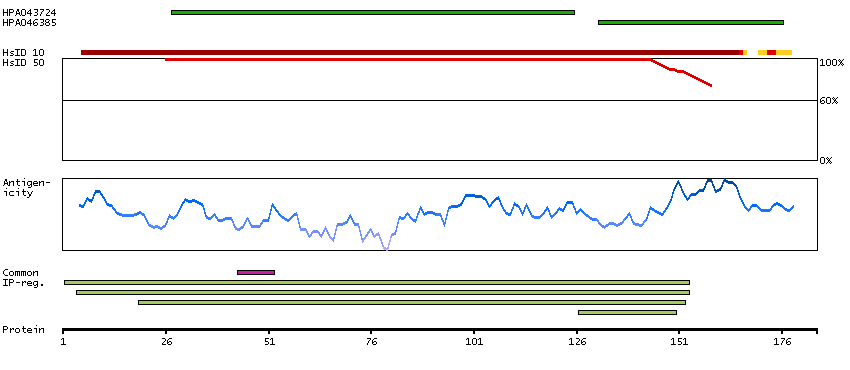
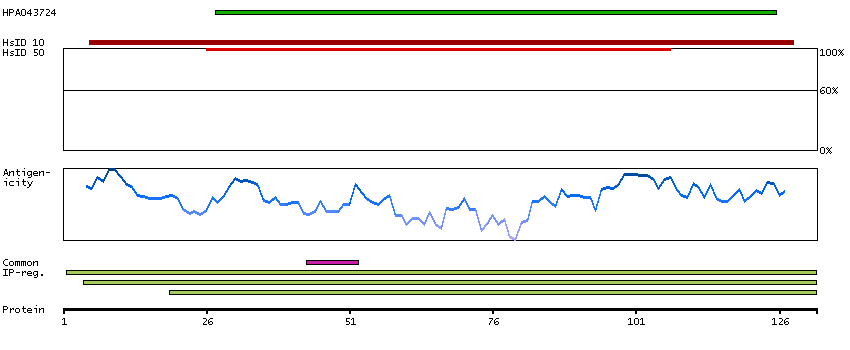
Ribosomes, the organelles that catalyze protein synthesis, consist of a small 40S subunit and a large 60S subunit. Together these subunits are composed of 4 RNA species and approximately 80 structurally distinct proteins. This gene encodes a ribosomal protein that is a component of the 60S subunit. The protein belongs to the L22P family of ribosomal proteins. It is located in the cytoplasm. This gene has been referred to as rpL23 because the encoded protein shares amino acid identity with ribosomal protein L23 from Halobacterium marismortui; however, its official symbol is RPL17. As is typical for genes encoding ribosomal proteins, there are multiple processed pseudogenes of this gene dispersed through the genome. Alternative splicing results in multiple transcript variants. Read-through transcription also exists between this gene and the neighboring downstream C18orf32 (chromosome 18 open reading frame 32) gene. [provided by RefSeq, Dec 2010]