We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
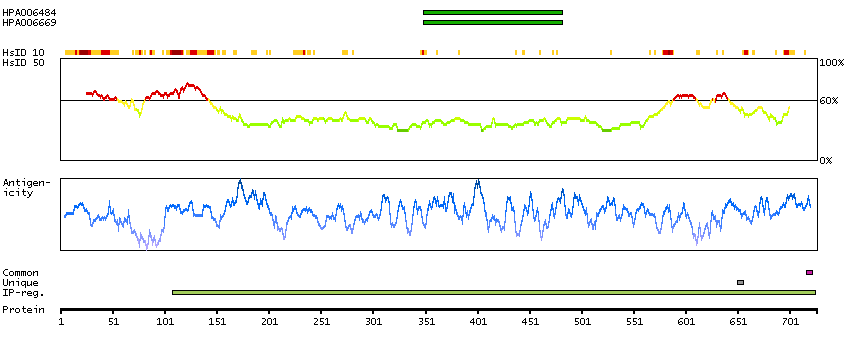
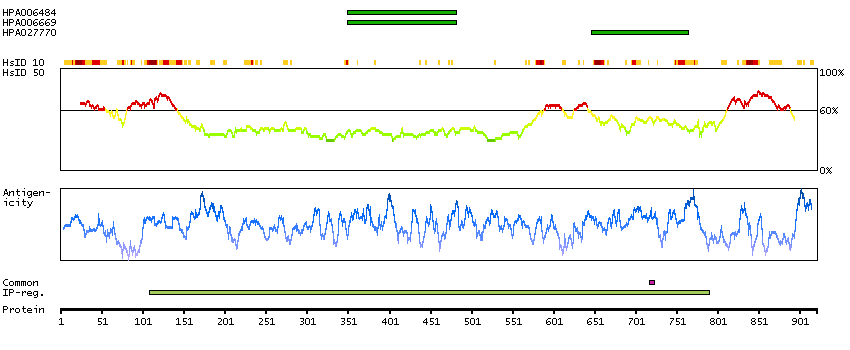
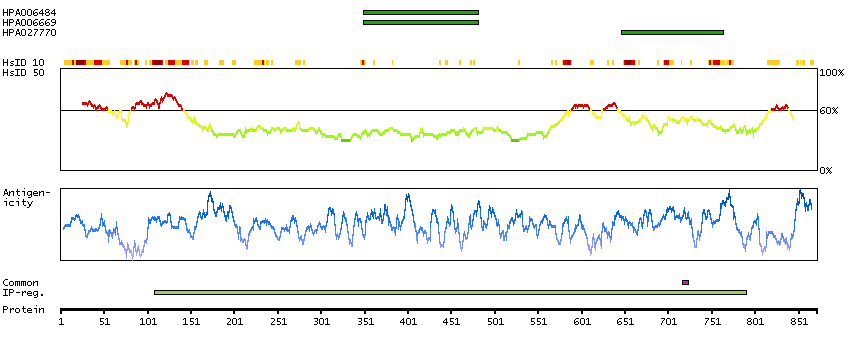
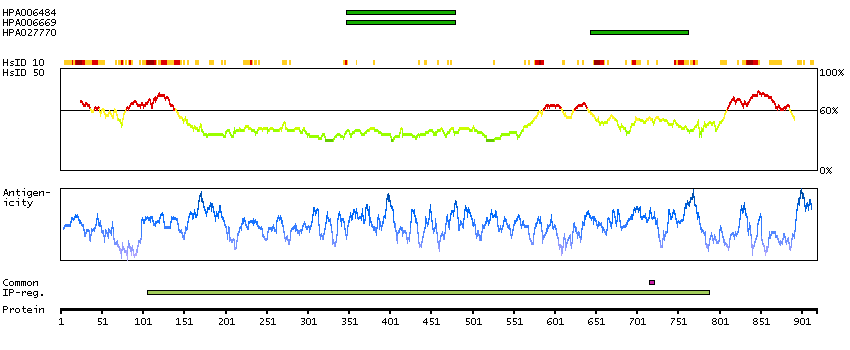
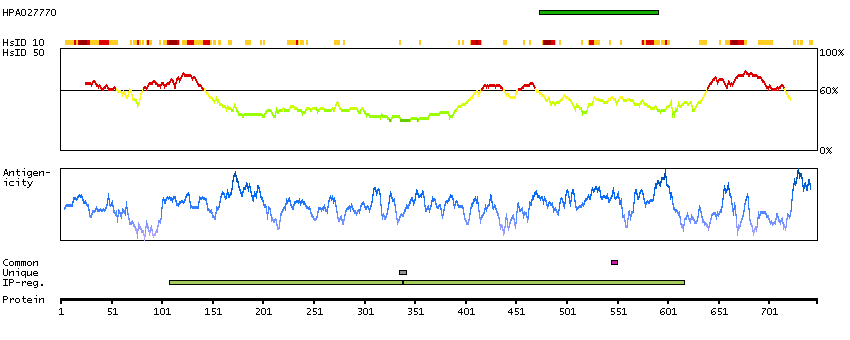
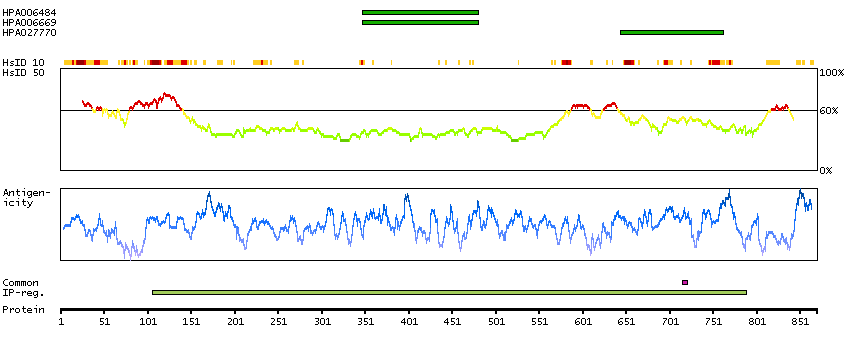
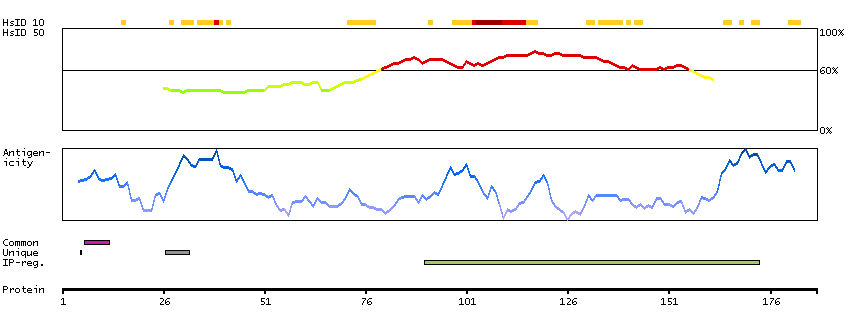
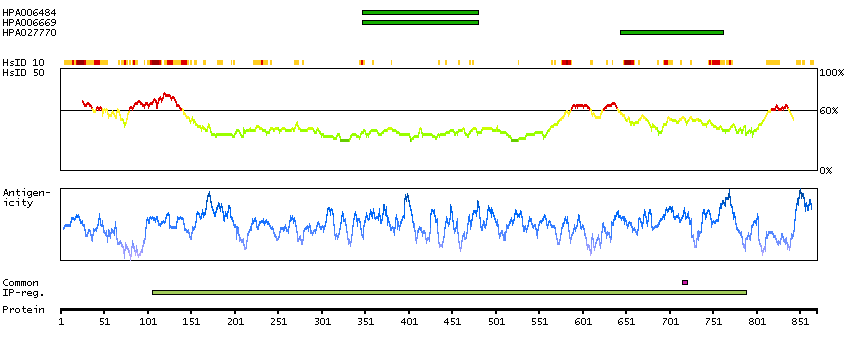
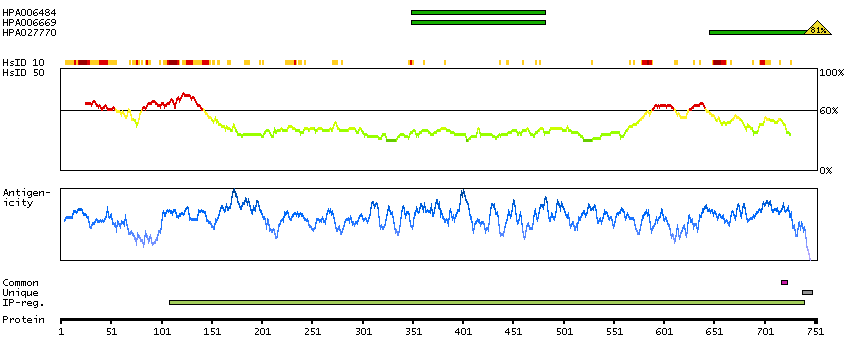
This gene encodes a transcriptional repressor that interacts with several members of the BCL2 family of proteins. Overexpression of this protein induces apoptosis, which can be suppressed by co-expression of BCL2 proteins. The protein localizes to dot-like structures throughout the nucleus, and redistributes to a zone near the nuclear envelope in cells undergoing apoptosis. Multiple transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jul 2008]
Predicted intracellular proteins Plasma proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003677 [DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0006351 [transcription, DNA-templated] GO:0006915 [apoptotic process] GO:0016607 [nuclear speck] GO:0043065 [positive regulation of apoptotic process] GO:0043620 [regulation of DNA-templated transcription in response to stress] GO:0044822 [poly(A) RNA binding] GO:0045892 [negative regulation of transcription, DNA-templated] GO:2000144 [positive regulation of DNA-templated transcription, initiation] GO:2001022 [positive regulation of response to DNA damage stimulus] GO:2001244 [positive regulation of intrinsic apoptotic signaling pathway]
Predicted intracellular proteins Plasma proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003677 [DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0006351 [transcription, DNA-templated] GO:0006915 [apoptotic process] GO:0016607 [nuclear speck] GO:0043065 [positive regulation of apoptotic process] GO:0043620 [regulation of DNA-templated transcription in response to stress] GO:0044822 [poly(A) RNA binding] GO:0045892 [negative regulation of transcription, DNA-templated] GO:2000144 [positive regulation of DNA-templated transcription, initiation] GO:2001022 [positive regulation of response to DNA damage stimulus] GO:2001244 [positive regulation of intrinsic apoptotic signaling pathway]
Predicted intracellular proteins Plasma proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003677 [DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0006351 [transcription, DNA-templated] GO:0006915 [apoptotic process] GO:0016607 [nuclear speck] GO:0043065 [positive regulation of apoptotic process] GO:0043620 [regulation of DNA-templated transcription in response to stress] GO:0044822 [poly(A) RNA binding] GO:0045892 [negative regulation of transcription, DNA-templated] GO:2000144 [positive regulation of DNA-templated transcription, initiation] GO:2001022 [positive regulation of response to DNA damage stimulus] GO:2001244 [positive regulation of intrinsic apoptotic signaling pathway]
Predicted intracellular proteins Plasma proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003677 [DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0006351 [transcription, DNA-templated] GO:0006915 [apoptotic process] GO:0016607 [nuclear speck] GO:0043065 [positive regulation of apoptotic process] GO:0043620 [regulation of DNA-templated transcription in response to stress] GO:0044822 [poly(A) RNA binding] GO:0045892 [negative regulation of transcription, DNA-templated] GO:2000144 [positive regulation of DNA-templated transcription, initiation] GO:2001022 [positive regulation of response to DNA damage stimulus] GO:2001244 [positive regulation of intrinsic apoptotic signaling pathway]
Predicted intracellular proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005634 [nucleus] GO:0006915 [apoptotic process] GO:0045892 [negative regulation of transcription, DNA-templated] GO:2001022 [positive regulation of response to DNA damage stimulus]
Predicted intracellular proteins Plasma proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003677 [DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005737 [cytoplasm] GO:0006351 [transcription, DNA-templated] GO:0006915 [apoptotic process] GO:0016607 [nuclear speck] GO:0043065 [positive regulation of apoptotic process] GO:0043620 [regulation of DNA-templated transcription in response to stress] GO:0044822 [poly(A) RNA binding] GO:0045892 [negative regulation of transcription, DNA-templated] GO:2000144 [positive regulation of DNA-templated transcription, initiation] GO:2001022 [positive regulation of response to DNA damage stimulus] GO:2001244 [positive regulation of intrinsic apoptotic signaling pathway]