We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
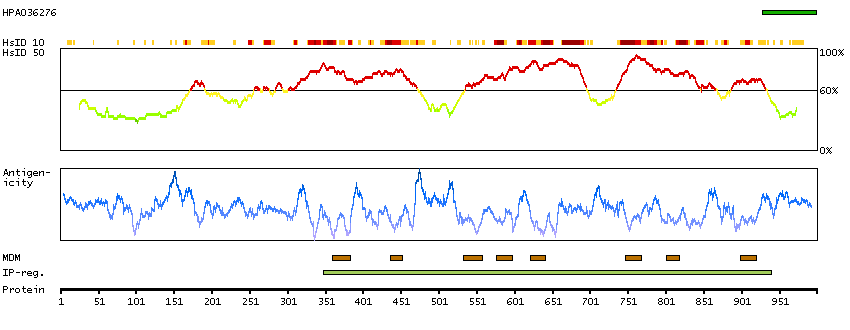
ANO2 belongs to a family of calcium-activated chloride channels (CaCCs) (reviewed by Hartzell et al., 2009 [PubMed 19015192]).[supplied by OMIM, Jan 2011]