We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
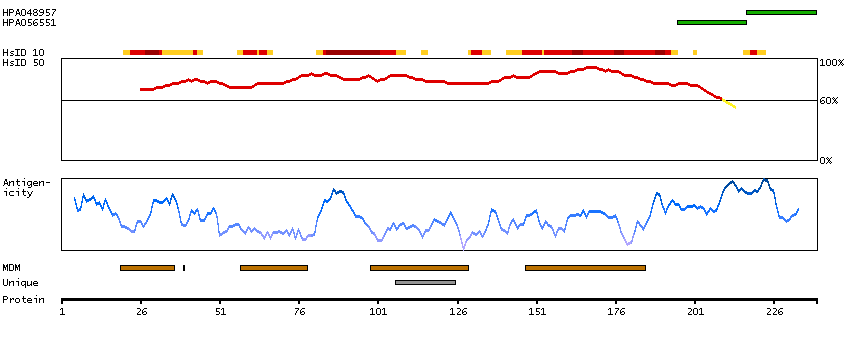
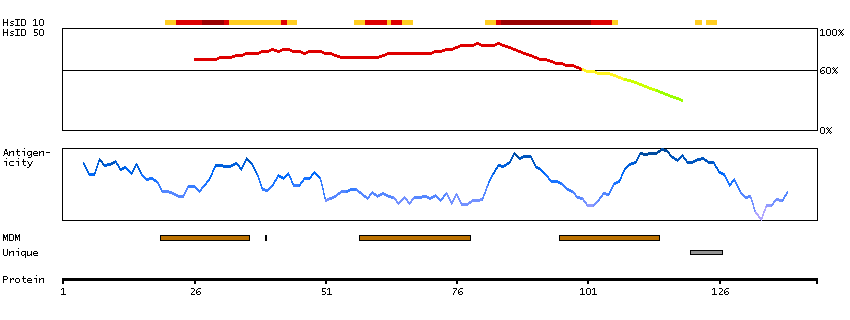
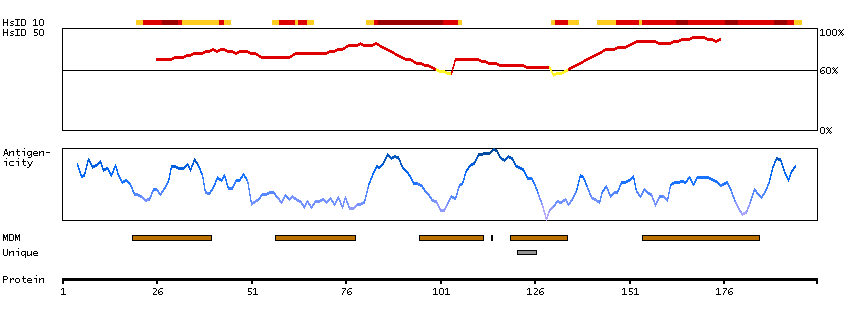
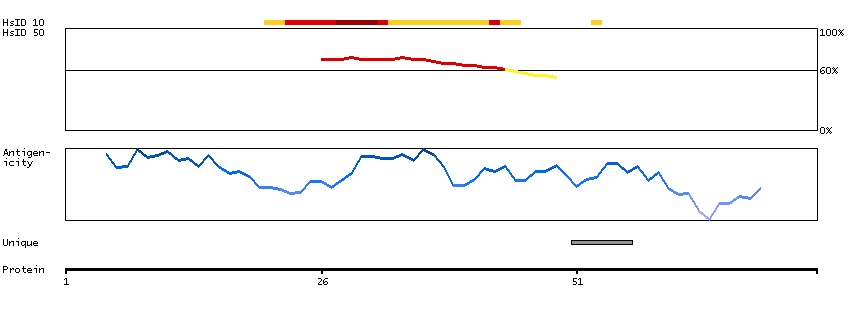
Proteins that are unfolded or misfolded in the endoplasmic reticulum (ER) must be refolded or degraded to maintain the homeostasis of the ER. DERL2 is involved in the degradation of misfolded glycoproteins in the ER (Oda et al., 2006 [PubMed 16449189]).[supplied by OMIM, Mar 2008]