We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
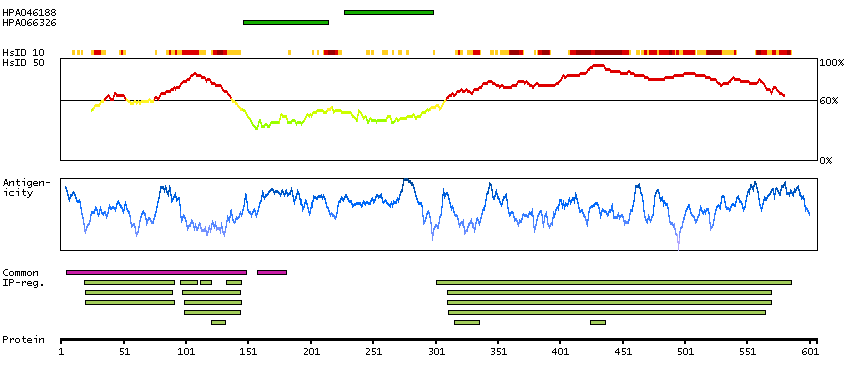
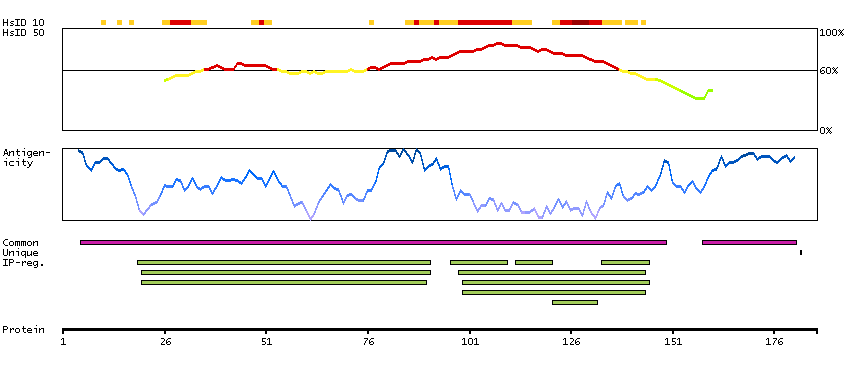
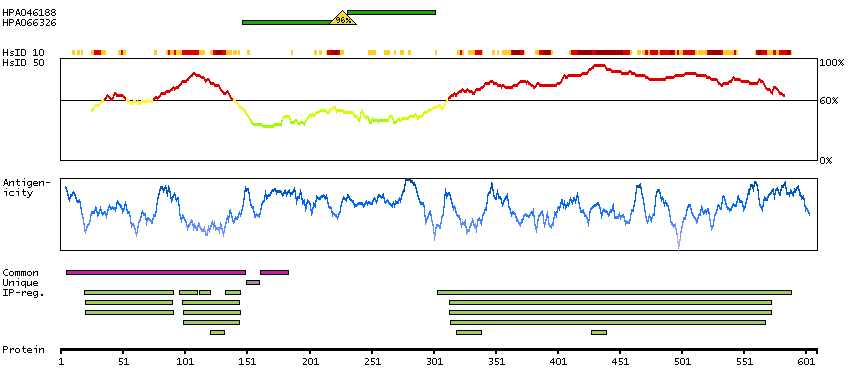
This proto-oncogene belongs to the RAF subfamily of the Ser/Thr protein kinase family, and maybe involved in cell growth and development. Alternatively spliced transcript variants encoding different isoforms have been found for this gene.[provided by RefSeq, Jan 2012]
Enzymes ENZYME proteins Transferases Kinases TKL Ser/Thr protein kinases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000165 [MAPK cascade] GO:0000186 [activation of MAPKK activity] GO:0004672 [protein kinase activity] GO:0004674 [protein serine/threonine kinase activity] GO:0005057 [receptor signaling protein activity] GO:0005515 [protein binding] GO:0005524 [ATP binding] GO:0005575 [cellular_component] GO:0005829 [cytosol] GO:0006464 [cellular protein modification process] GO:0006468 [protein phosphorylation] GO:0007165 [signal transduction] GO:0007173 [epidermal growth factor receptor signaling pathway] GO:0007264 [small GTPase mediated signal transduction] GO:0007265 [Ras protein signal transduction] GO:0007411 [axon guidance] GO:0008286 [insulin receptor signaling pathway] GO:0008543 [fibroblast growth factor receptor signaling pathway] GO:0032006 [regulation of TOR signaling] GO:0032434 [regulation of proteasomal ubiquitin-dependent protein catabolic process] GO:0033138 [positive regulation of peptidyl-serine phosphorylation] GO:0035556 [intracellular signal transduction] GO:0038095 [Fc-epsilon receptor signaling pathway] GO:0043066 [negative regulation of apoptotic process] GO:0045087 [innate immune response] GO:0046872 [metal ion binding] GO:0048010 [vascular endothelial growth factor receptor signaling pathway] GO:0048011 [neurotrophin TRK receptor signaling pathway]
Enzymes ENZYME proteins Transferases Kinases TKL Ser/Thr protein kinases Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000165 [MAPK cascade] GO:0000186 [activation of MAPKK activity] GO:0004672 [protein kinase activity] GO:0004674 [protein serine/threonine kinase activity] GO:0005057 [receptor signaling protein activity] GO:0005515 [protein binding] GO:0005524 [ATP binding] GO:0005575 [cellular_component] GO:0005829 [cytosol] GO:0006464 [cellular protein modification process] GO:0007165 [signal transduction] GO:0007173 [epidermal growth factor receptor signaling pathway] GO:0007264 [small GTPase mediated signal transduction] GO:0007265 [Ras protein signal transduction] GO:0007411 [axon guidance] GO:0008286 [insulin receptor signaling pathway] GO:0008543 [fibroblast growth factor receptor signaling pathway] GO:0032006 [regulation of TOR signaling] GO:0032434 [regulation of proteasomal ubiquitin-dependent protein catabolic process] GO:0033138 [positive regulation of peptidyl-serine phosphorylation] GO:0035556 [intracellular signal transduction] GO:0038095 [Fc-epsilon receptor signaling pathway] GO:0043066 [negative regulation of apoptotic process] GO:0045087 [innate immune response] GO:0046872 [metal ion binding] GO:0048010 [vascular endothelial growth factor receptor signaling pathway] GO:0048011 [neurotrophin TRK receptor signaling pathway]