We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
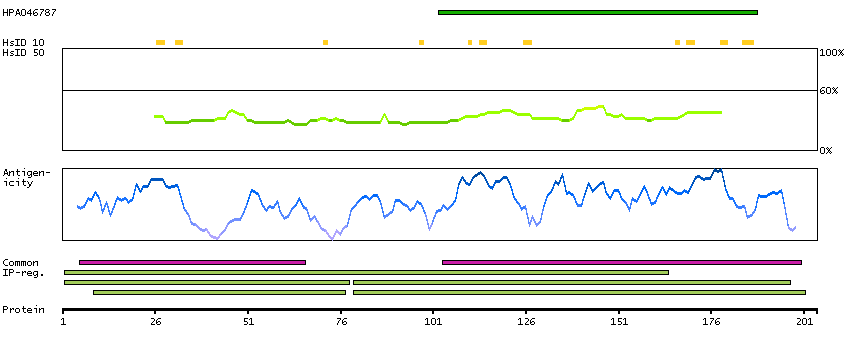
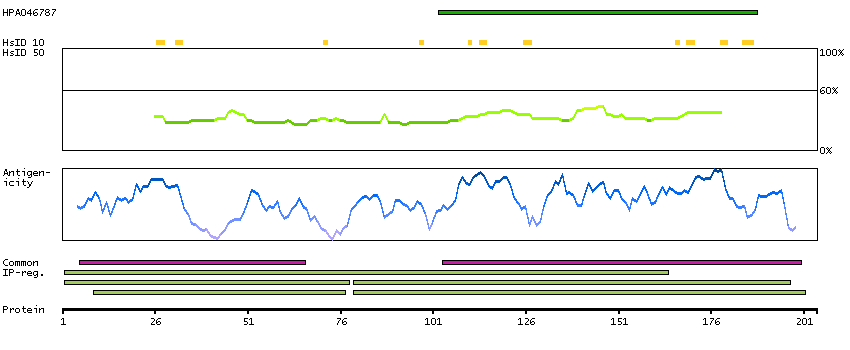
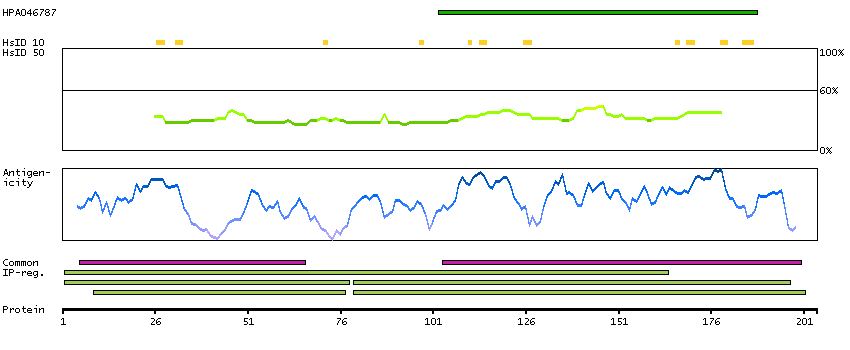
Q9Y535 [Direct mapping] DNA-directed RNA polymerase III subunit RPC8
Show all
MEMSAT3 predicted membrane proteins Predicted intracellular proteins RNA polymerase related proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0001056 [RNA polymerase III activity] GO:0003677 [DNA binding] GO:0003899 [DNA-directed RNA polymerase activity] GO:0005654 [nucleoplasm] GO:0005666 [DNA-directed RNA polymerase III complex] GO:0005813 [centrosome] GO:0005829 [cytosol] GO:0006139 [nucleobase-containing compound metabolic process] GO:0006351 [transcription, DNA-templated] GO:0006383 [transcription from RNA polymerase III promoter] GO:0006384 [transcription initiation from RNA polymerase III promoter] GO:0006385 [transcription elongation from RNA polymerase III promoter] GO:0006386 [termination of RNA polymerase III transcription] GO:0010467 [gene expression] GO:0032481 [positive regulation of type I interferon production] GO:0043231 [intracellular membrane-bounded organelle] GO:0045087 [innate immune response] GO:0051607 [defense response to virus]