We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
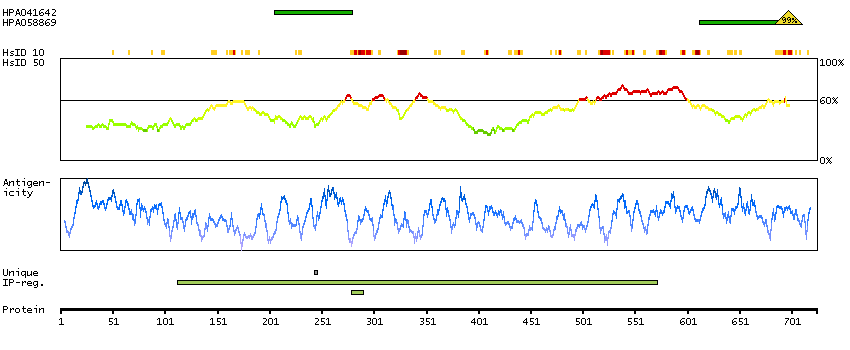
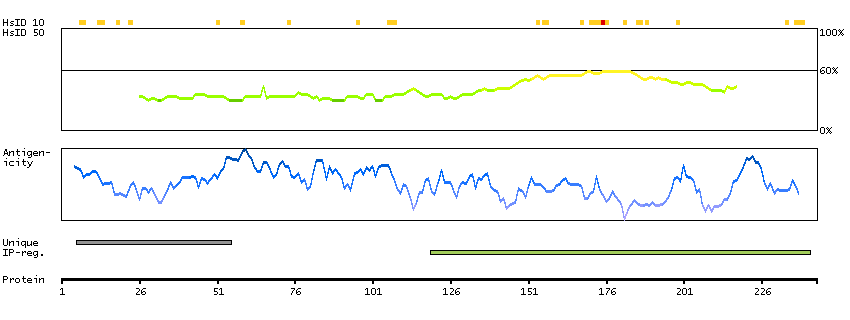
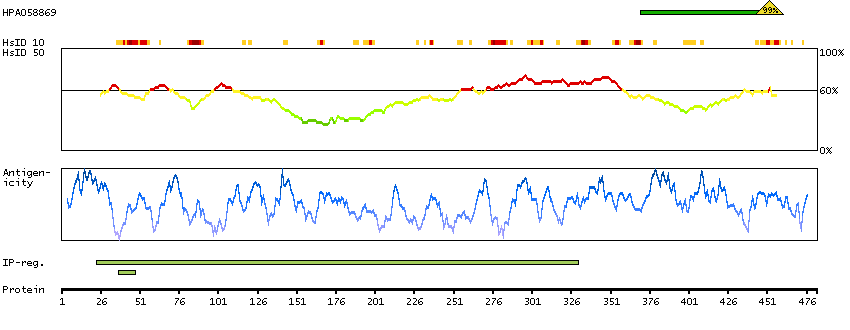
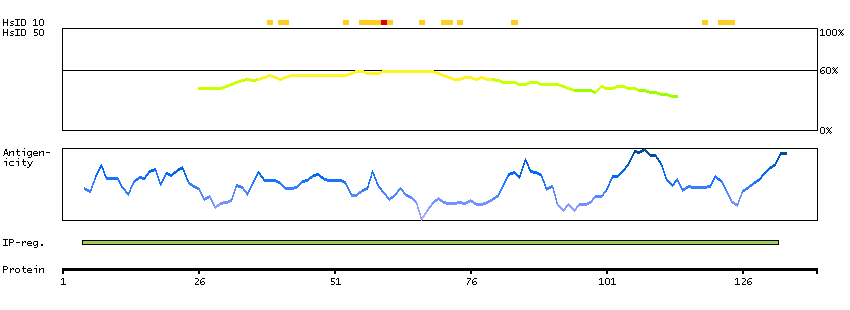
Acyl-CoA synthetase bubblegum family member 1 (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene possesses long-chain acyl-CoA synthetase activity. It is thought to play a central role in brain very long-chain fatty acids metabolism and myelinogenesis. [provided by RefSeq, Jul 2008]