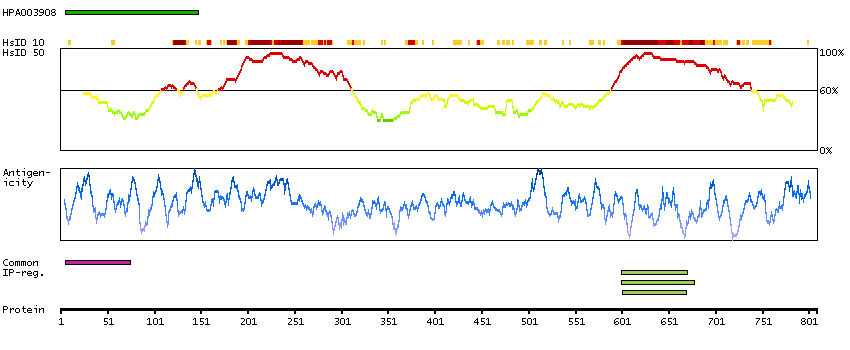
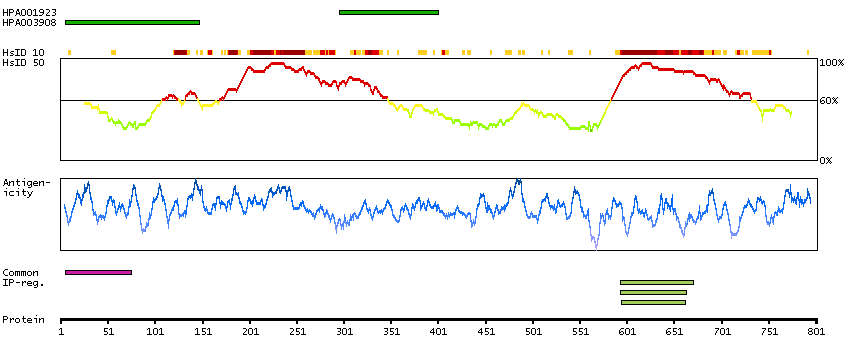
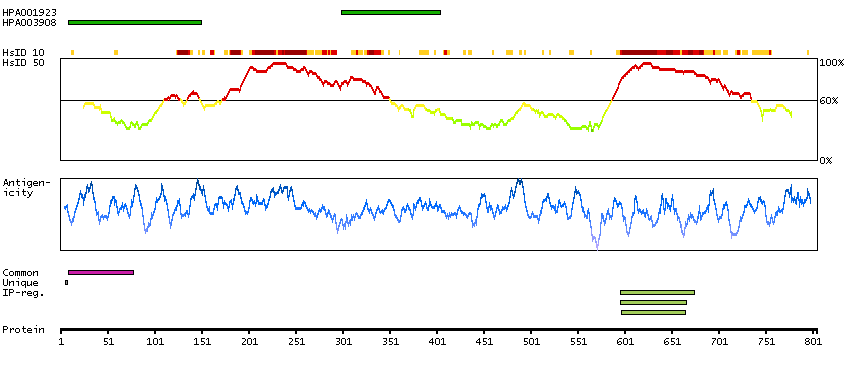
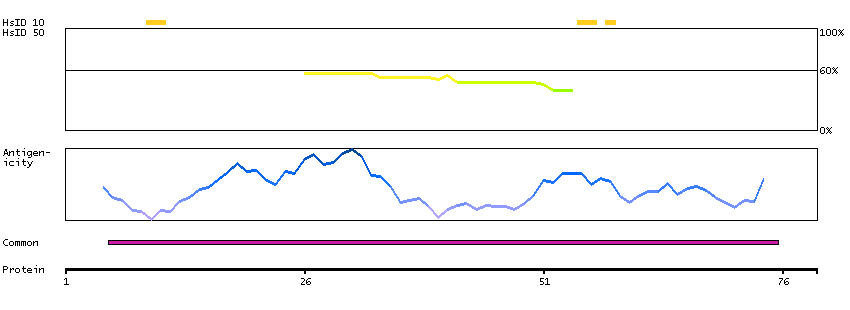
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
SRY (sex determining region Y)-box 6 (HGNC Symbol)
Entrez gene summary
This gene encodes a member of the D subfamily of sex determining region y-related transcription factors that are characterized by a conserved DNA-binding domain termed the high mobility group box and by their ability to bind the minor groove of DNA. The encoded protein is a transcriptional activator that is required for normal development of the central nervous system, chondrogenesis and maintenance of cardiac and skeletal muscle cells. The encoded protein interacts with other family members to cooperatively activate gene expression. Alternative splicing results in multiple transcript variants.[provided by RefSeq, Mar 2009]