We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
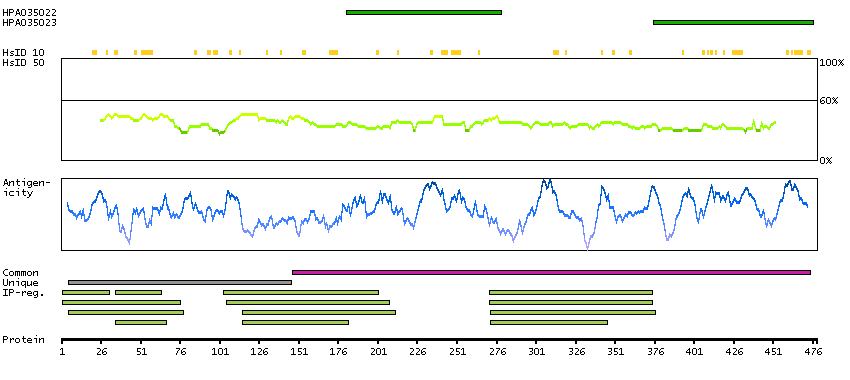
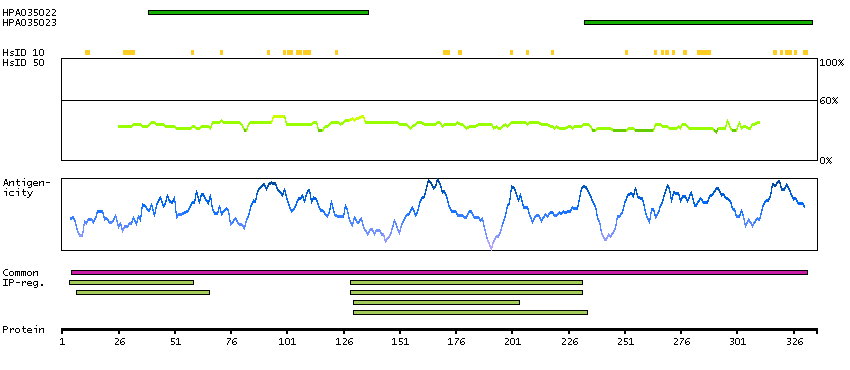
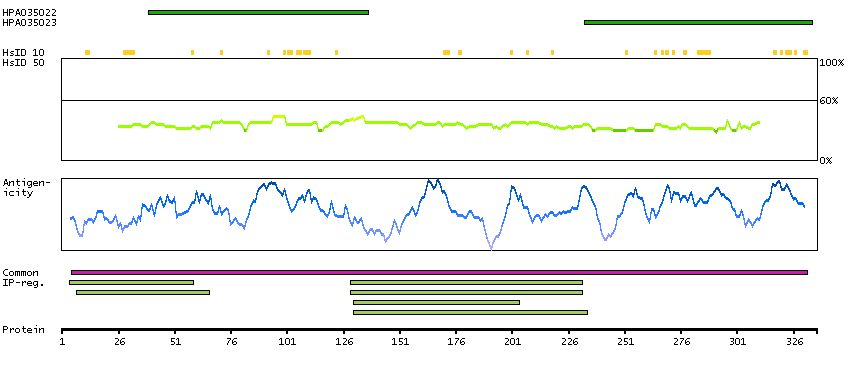
Ankyrin repeat and BTB (POZ) domain containing 1 (HGNC Symbol)
Entrez gene summary
This gene encodes a protein with an ankyrin repeat region and two BTB/POZ domains, which are thought to be involved in protein-protein interactions. Expression of this gene is activated by the phosphatase and tensin homolog, a tumor suppressor. Alternate splicing results in three transcript variants. [provided by RefSeq, Mar 2010]