We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
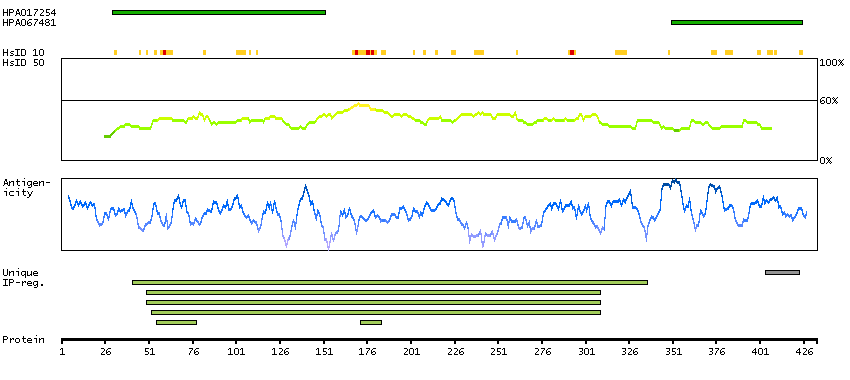
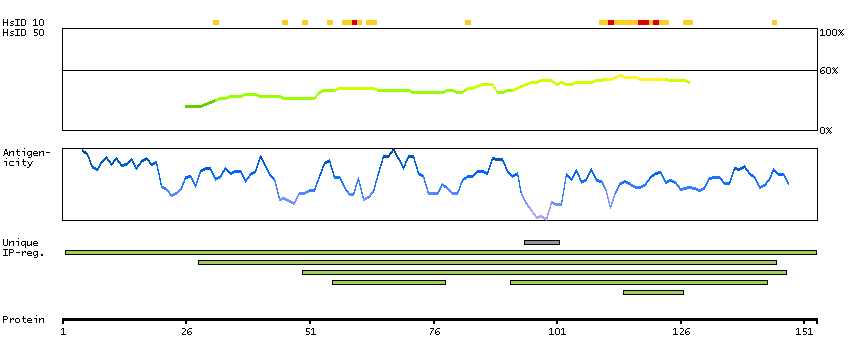
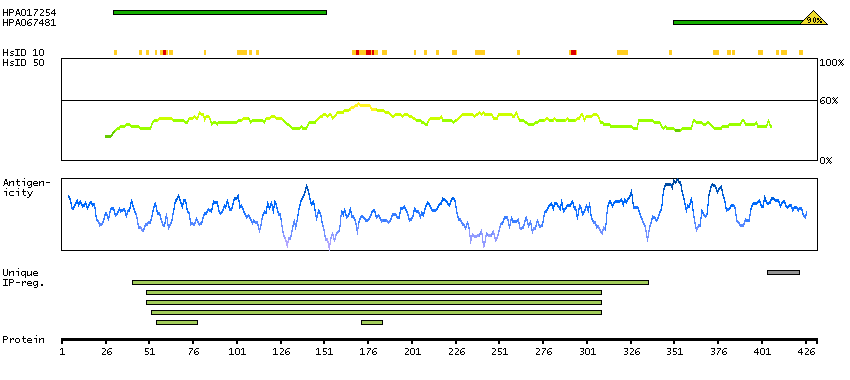
This gene, which encodes a member of the serine/threonine kinase family, regulates cell polarity and functions as a tumor suppressor. Mutations in this gene have been associated with Peutz-Jeghers syndrome, an autosomal dominant disorder characterized by the growth of polyps in the gastrointestinal tract, pigmented macules on the skin and mouth, and other neoplasms. Alternate transcriptional splice variants of this gene have been observed but have not been thoroughly characterized. [provided by RefSeq, Jul 2008]