We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
DNA (cytosine-5-)-methyltransferase 3 alpha (HGNC Symbol)
Entrez gene summary
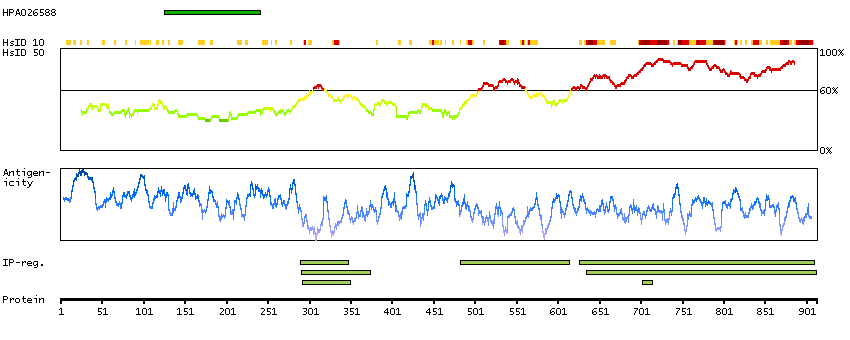
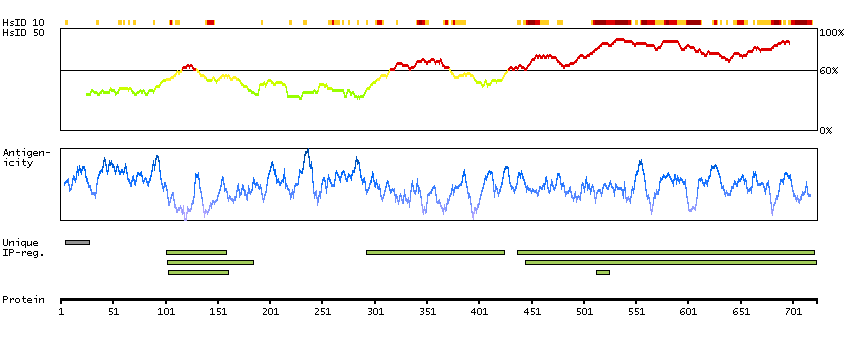
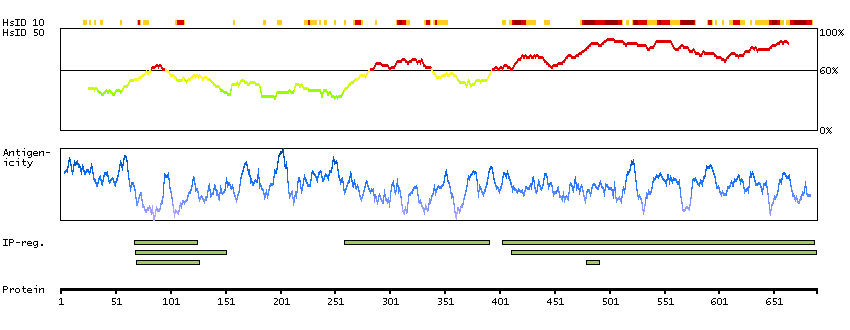
CpG methylation is an epigenetic modification that is important for embryonic development, imprinting, and X-chromosome inactivation. Studies in mice have demonstrated that DNA methylation is required for mammalian development. This gene encodes a DNA methyltransferase that is thought to function in de novo methylation, rather than maintenance methylation. The protein localizes to the cytoplasm and nucleus and its expression is developmentally regulated. Alternative splicing results in multiple transcript variants encoding different isoforms. [provided by RefSeq, Jul 2008]