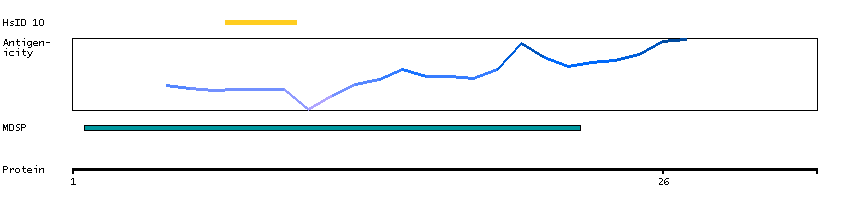
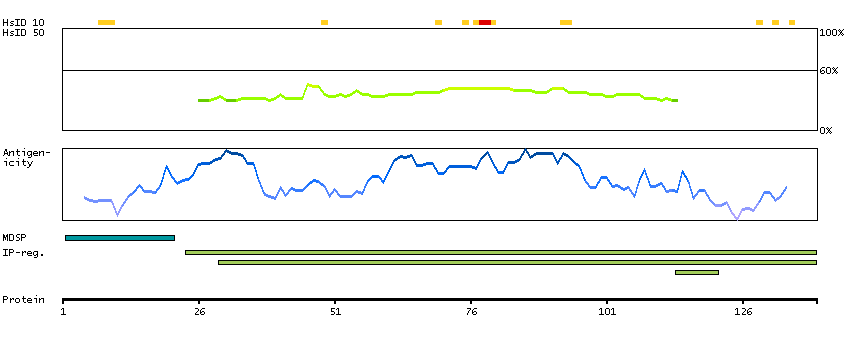
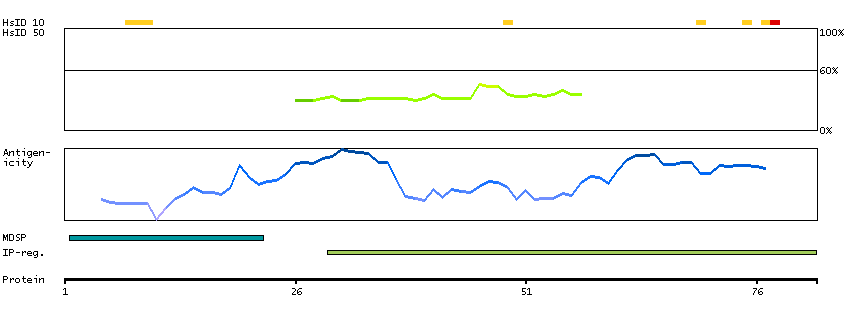
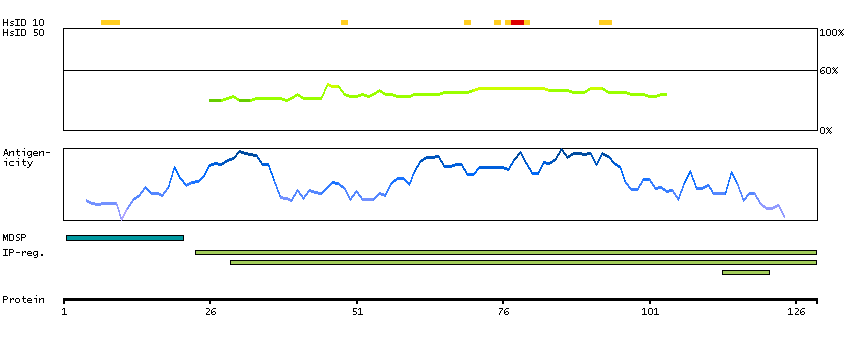
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
The protein encoded by this gene is a secreted chaperone that can under some stress conditions also be found in the cell cytosol. It has been suggested to be involved in several basic biological events such as cell death, tumor progression, and neurodegenerative disorders. Alternate splicing results in both coding and non-coding variants.[provided by RefSeq, May 2011]