We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
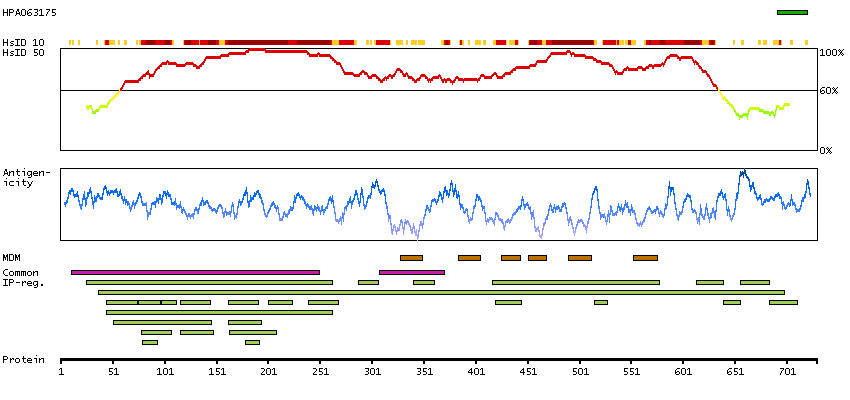
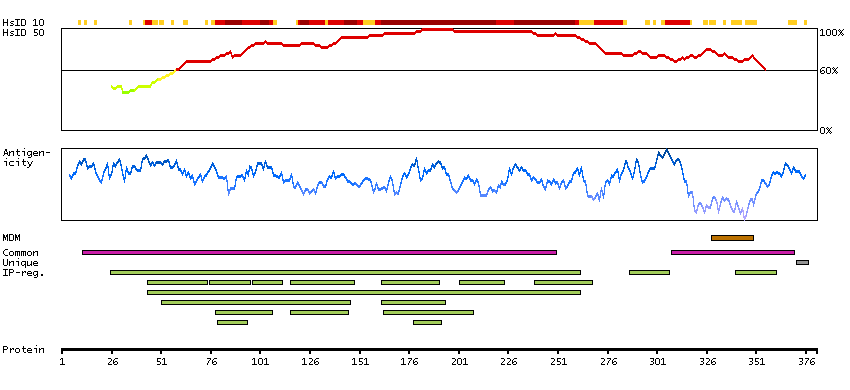
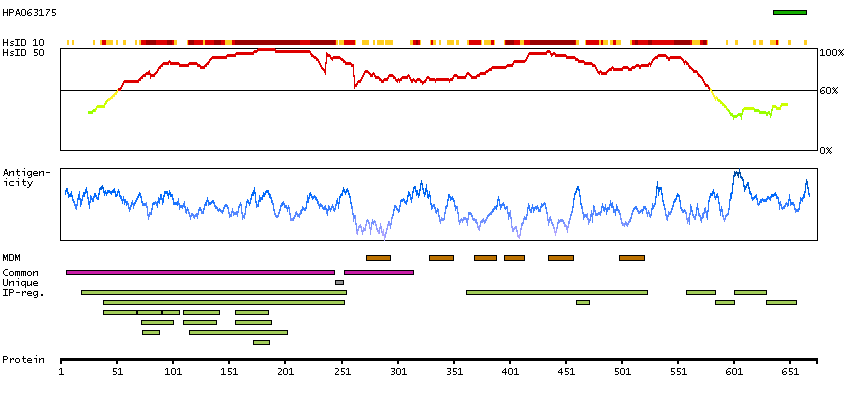
This gene is a member of the transient receptor family and the TrpV subfamily. The calcium-selective channel encoded by this gene has 6 transmembrane-spanning domains, multiple potential phosphorylation sites, an N-linked glycosylation site, and 5 ANK repeats. This protein forms homotetramers or heterotetramers and is activated by a low internal calcium level. [provided by RefSeq, Jul 2008]