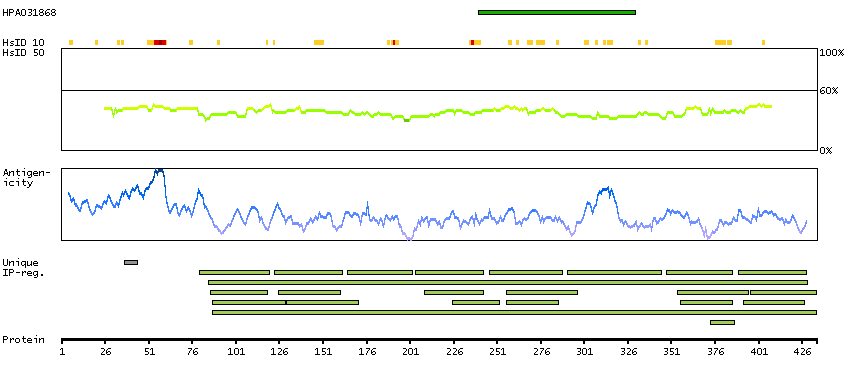
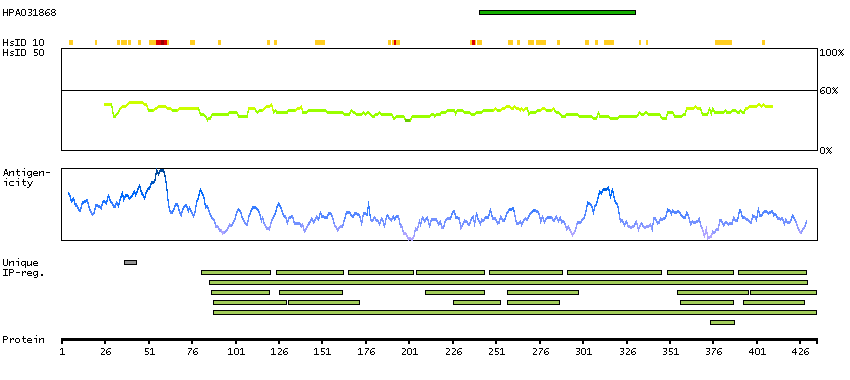
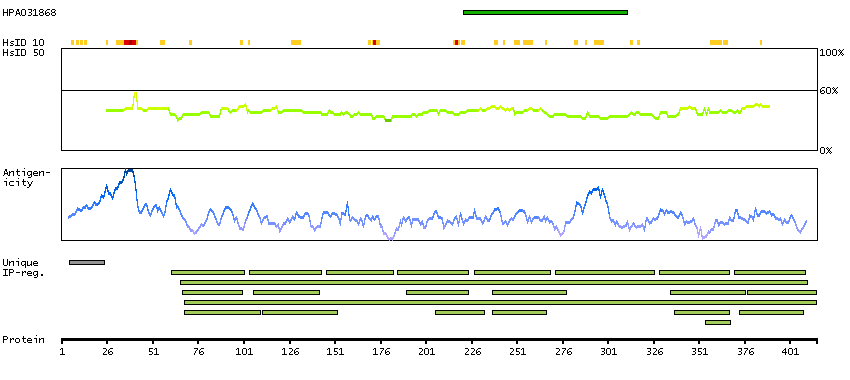
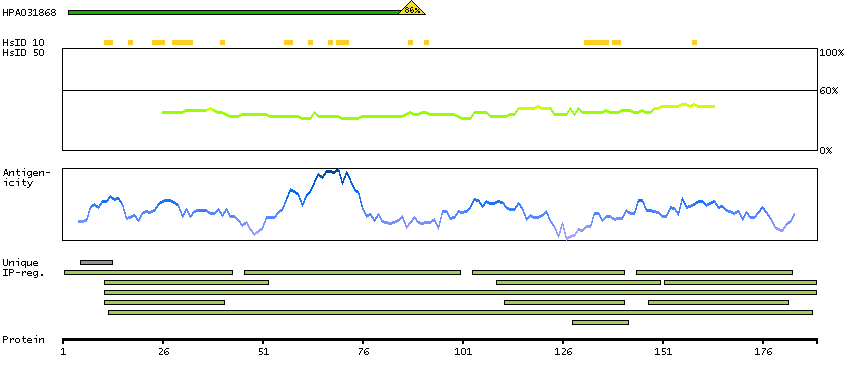
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Angio-associated, migratory cell protein (HGNC Symbol)
Entrez gene summary
The gene is a member of the immunoglobulin superfamily. The encoded protein is associated with angiogenesis, with potential roles in endothelial tube formation and the migration of endothelial cells. It may also regulate smooth muscle cell migration via the RhoA pathway. The encoded protein can bind to heparin and may mediate heparin-sensitive cell adhesion. [provided by RefSeq, Oct 2014]