We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
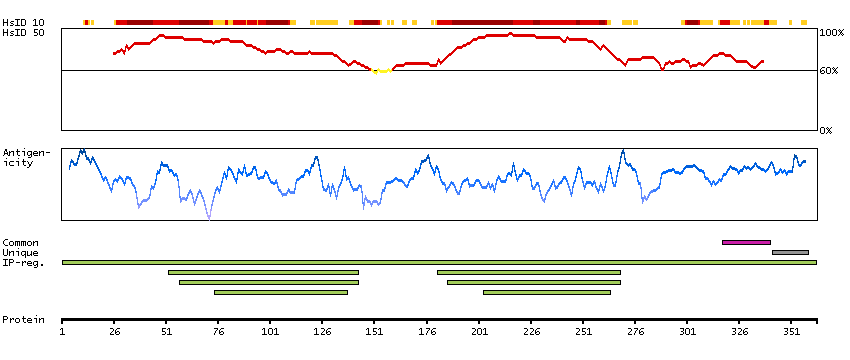
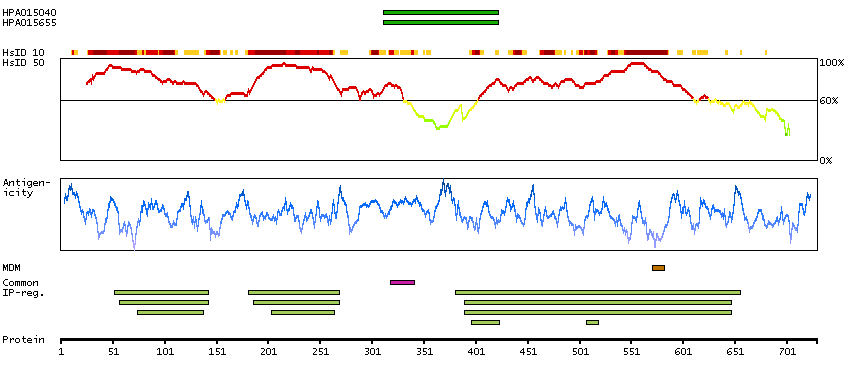
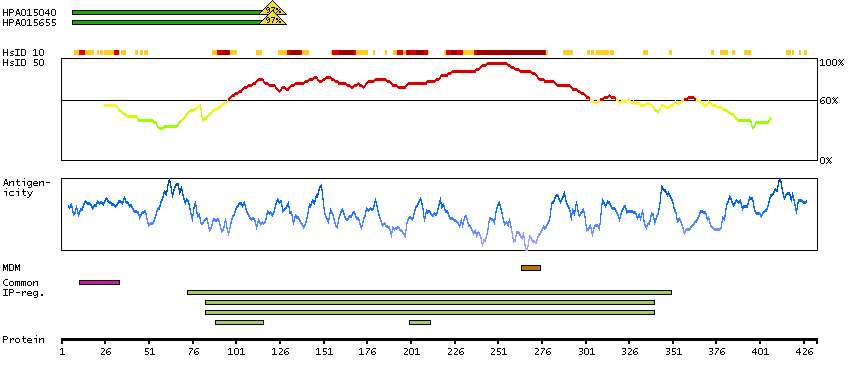
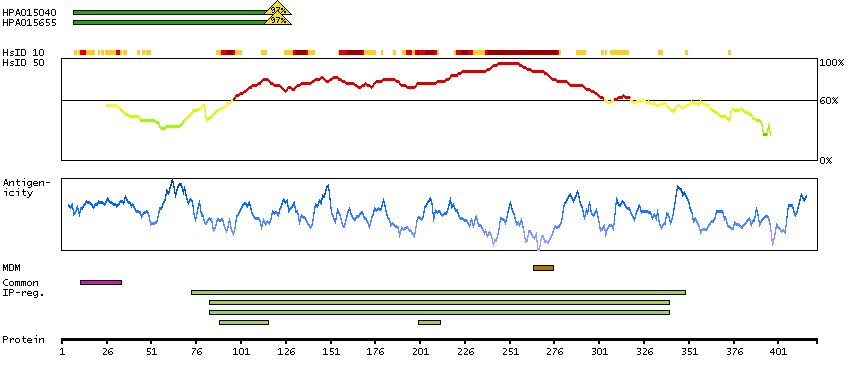
This gene encodes a member of the protein kinase superfamily and the doublecortin family. The protein encoded by this gene contains two N-terminal doublecortin domains, which bind microtubules and regulate microtubule polymerization, a C-terminal serine/threonine protein kinase domain, which shows substantial homology to Ca2+/calmodulin-dependent protein kinase, and a serine/proline-rich domain in between the doublecortin and the protein kinase domains, which mediates multiple protein-protein interactions. The microtubule-polymerizing activity of the encoded protein is independent of its protein kinase activity. The encoded protein is involved in several different cellular processes, including neuronal migration, retrograde transport, neuronal apoptosis and neurogenesis. This gene is up-regulated by brain-derived neurotrophic factor and associated with memory and general cognitive abilities. Multiple transcript variants generated by two alternative promoter usage and alternative splicing have been reported, but the full-length nature and biological validity of some variants have not been defined. These variants encode different isoforms, which are differentially expressed and have different kinase activities.[provided by RefSeq, Sep 2010]