We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
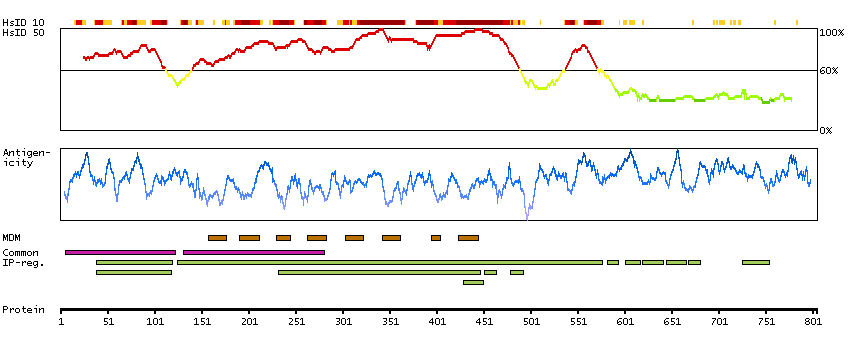
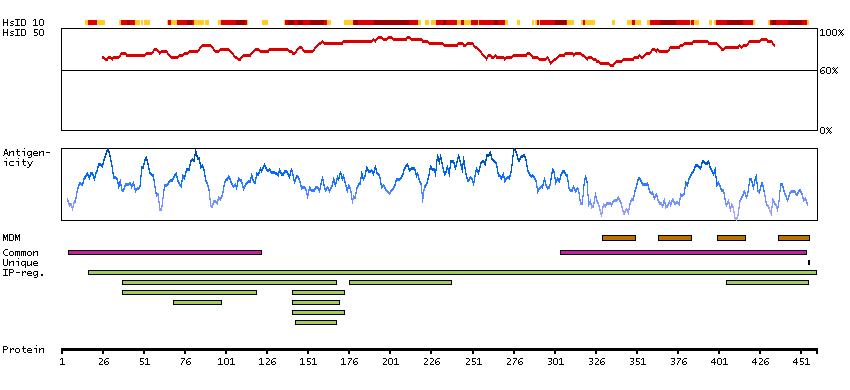
This gene encodes a member of the canonical subfamily of transient receptor potential cation channels. The encoded protein forms a non-selective calcium-permeable cation channel that is activated by Gq-coupled receptors and tyrosine kinases, and plays a role in multiple processes including endothelial permeability, vasodilation, neurotransmitter release and cell proliferation. Single nucleotide polymorphisms in this gene may be associated with generalized epilepsy with photosensitivity. Alternatively spliced transcript variants encoding multiple isoforms have been observed for this gene. [provided by RefSeq, Aug 2011]