We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
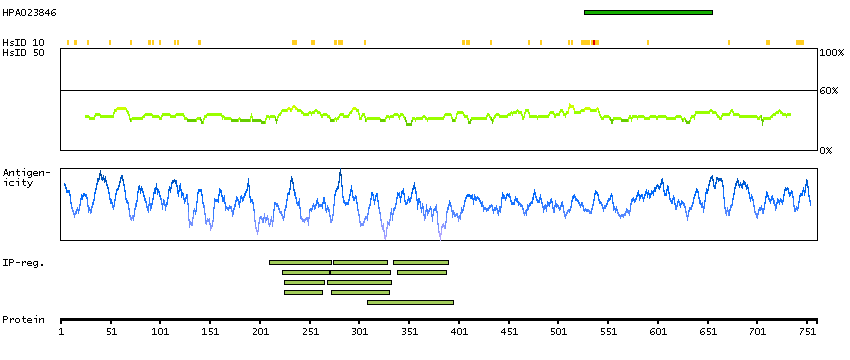
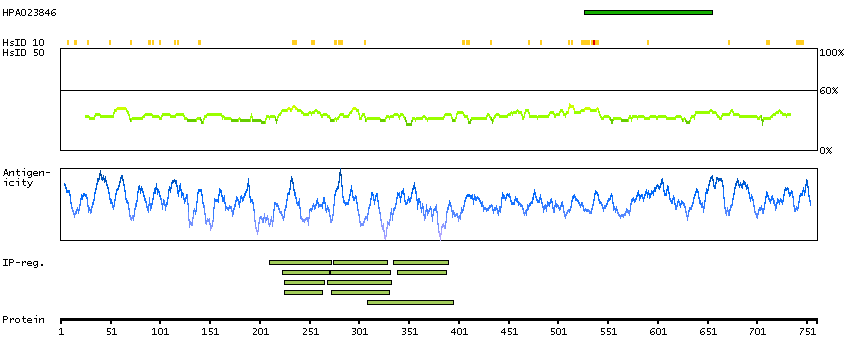
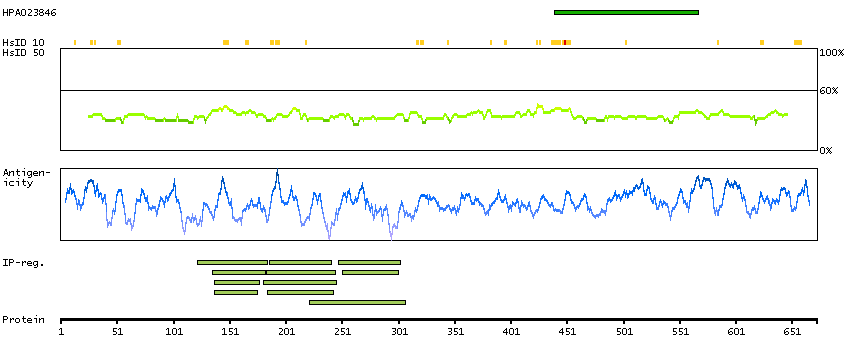
Cyclin D binding myb-like transcription factor 1 (HGNC Symbol)
Entrez gene summary
This gene encodes a transcription factor that contains a cyclin D-binding domain, three central Myb-like repeats, and two flanking acidic transactivation domains at the N- and C-termini. The encoded protein is induced by the oncogenic Ras signaling pathway and functions as a tumor suppressor by activating the transcription of ARF and thus the ARF-p53 pathway to arrest cell growth or induce apoptosis. It also activates the transcription of aminopeptidase N and may play a role in hematopoietic cell differentiation. The transcriptional activity of this protein is regulated by binding of D-cyclins. This gene is hemizygously deleted in approximately 40% of human non-small-cell lung cancer and is a potential prognostic and gene-therapy target for non-small-cell lung cancer. Multiple transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Dec 2008]
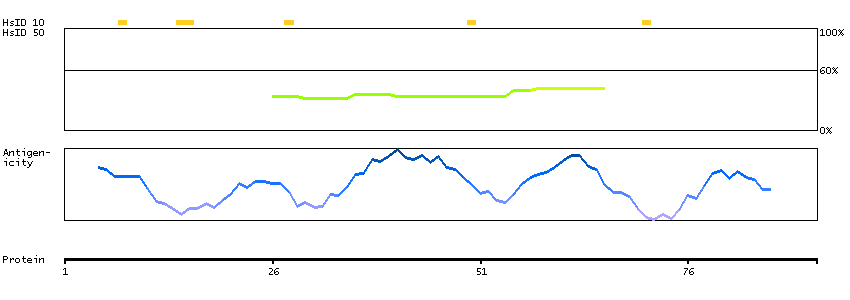
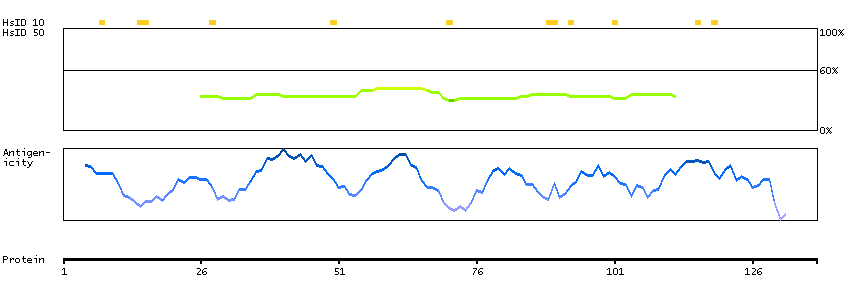
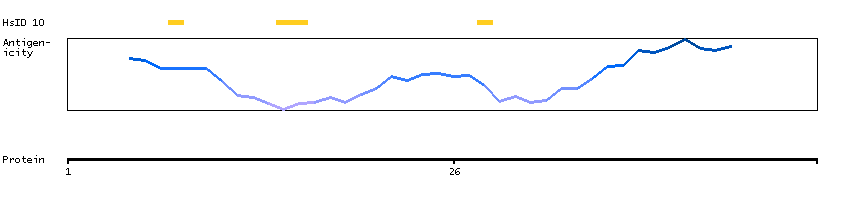
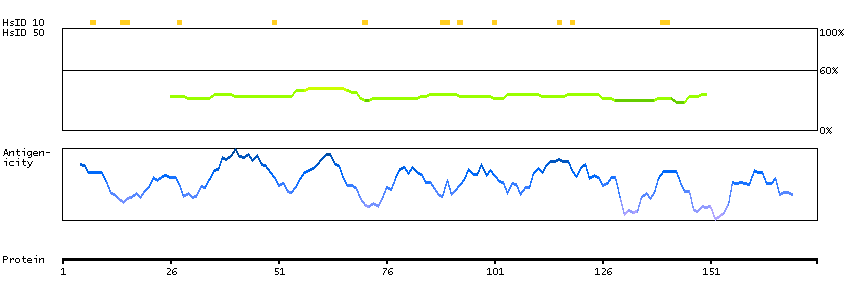
Predicted intracellular proteins Transcription factors Helix-turn-helix domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
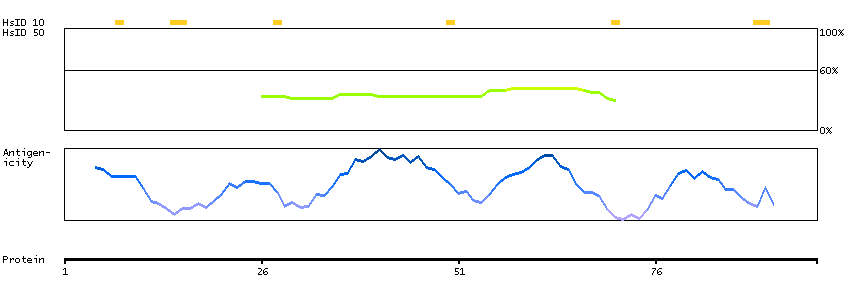
Predicted intracellular proteins Transcription factors Helix-turn-helix domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
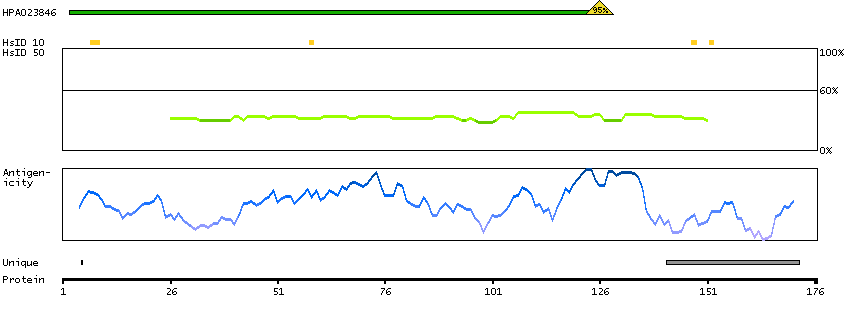
Predicted intracellular proteins Transcription factors Helix-turn-helix domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)