We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
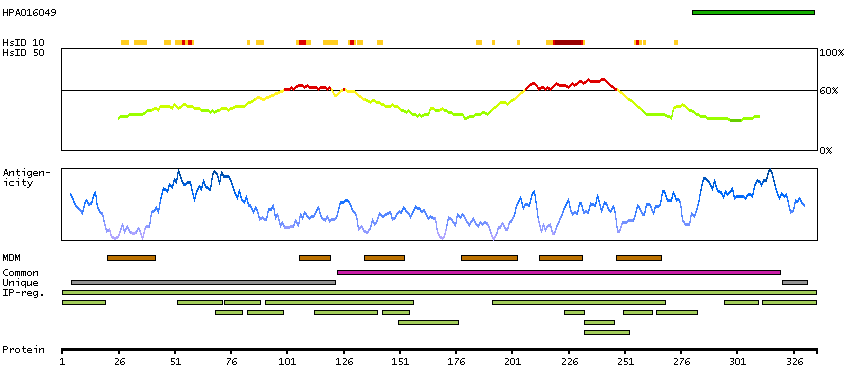
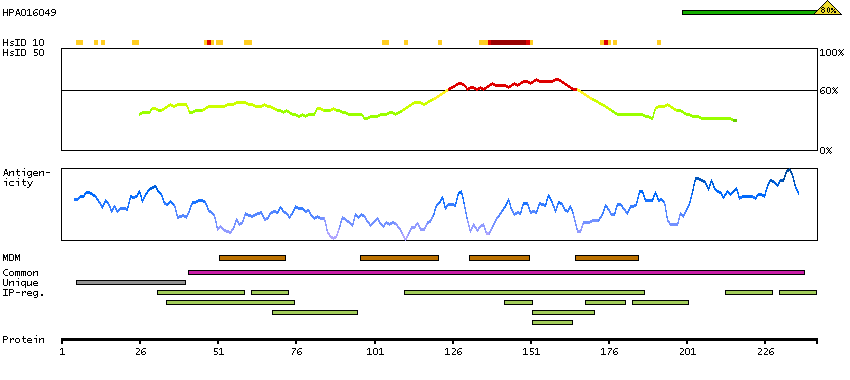
Potassium channel, two pore domain subfamily K, member 1 (HGNC Symbol)
Entrez gene summary
This gene encodes one of the members of the superfamily of potassium channel proteins containing two pore-forming P domains. The product of this gene has not been shown to be a functional channel, however, it may require other non-pore-forming proteins for activity. [provided by RefSeq, Jul 2008]