We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
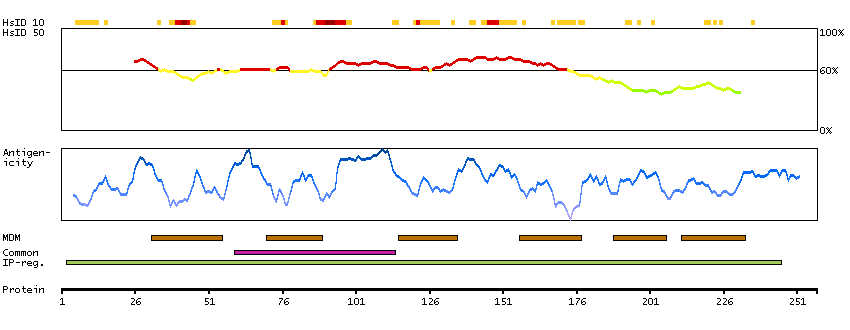
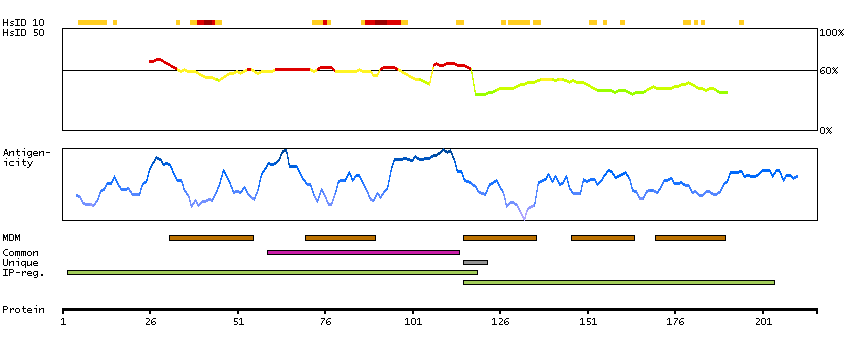
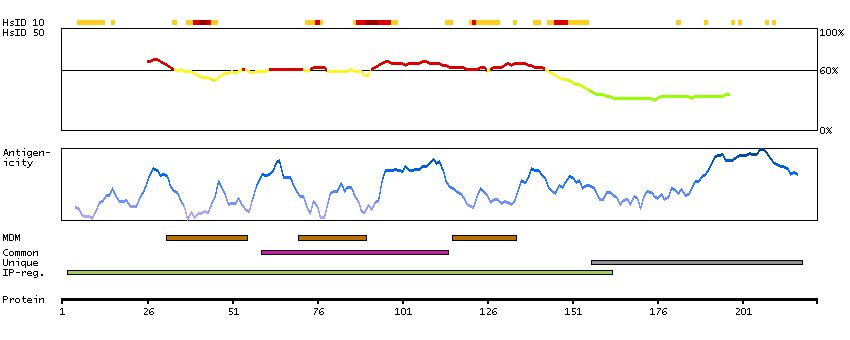
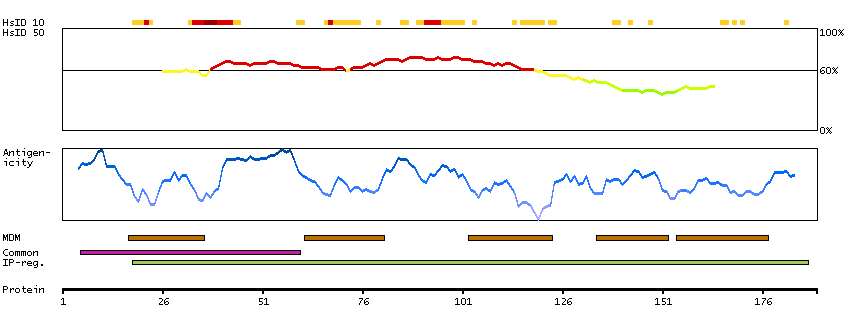
This gene encodes a multi-pass transmembrane protein that is a functional component of the gamma-secretase complex, which also contains presenilin and nicastrin. This protein represents a stabilizing cofactor for the presenilin holoprotein in the complex. The gamma-secretase complex catalyzes the cleavage of integral proteins such as notch receptors and beta-amyloid precursor protein. [provided by RefSeq, Sep 2011]