We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
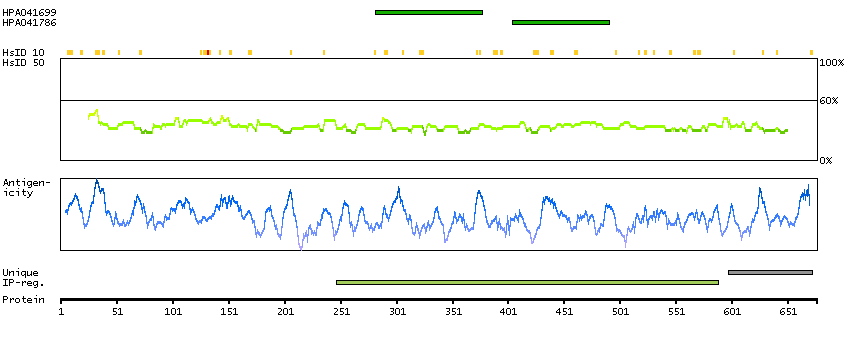
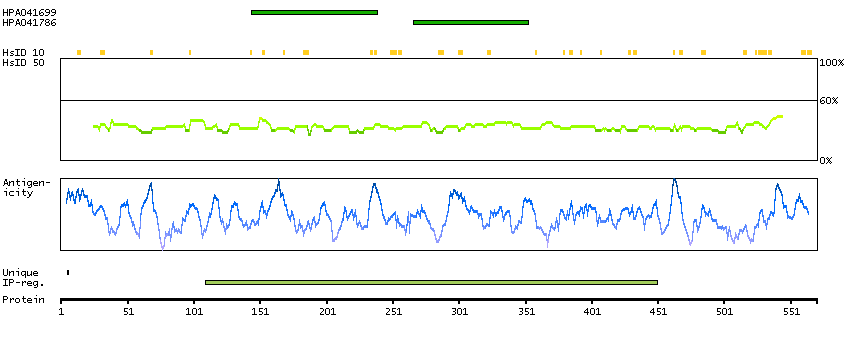
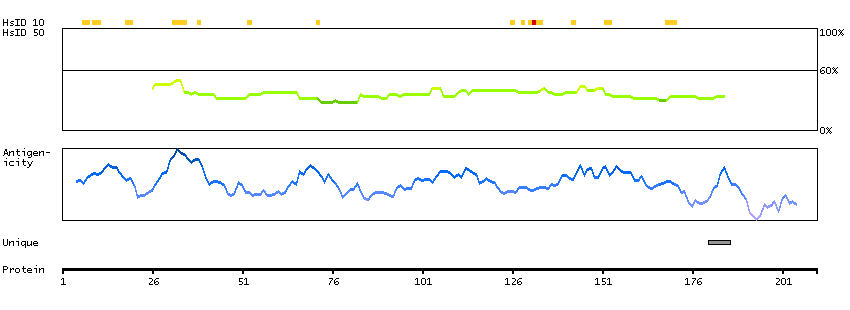
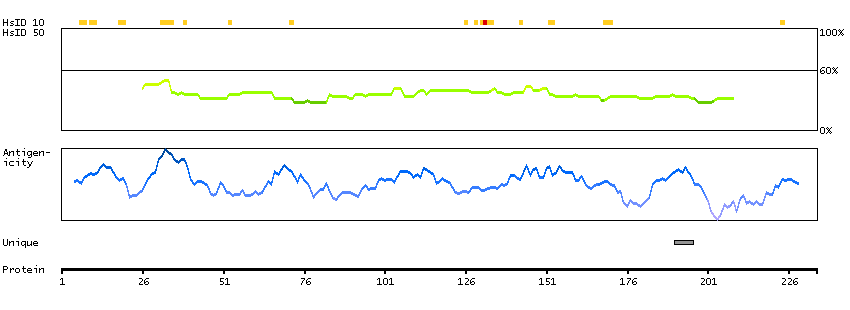
TCF25 is a member of the basic helix-loop-helix (bHLH) family of transcription factors that are important in embryonic development (Steen and Lindholm, 2008 [PubMed 18068114]).[supplied by OMIM, Sep 2008]
Predicted intracellular proteins Transcription factors Yet undefined DNA-binding domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0006351 [transcription, DNA-templated] GO:0007507 [heart development]