We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Polymerase (RNA) II (DNA directed) polypeptide D (HGNC Symbol)
Entrez gene summary
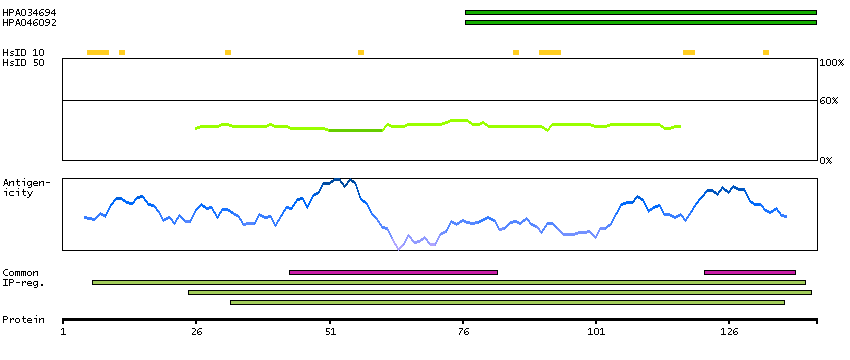
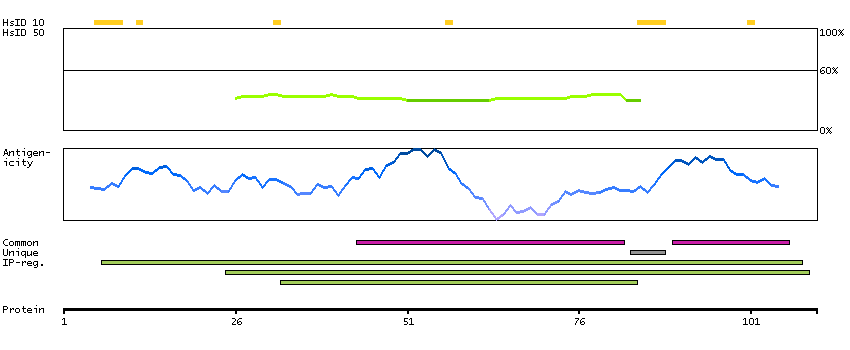
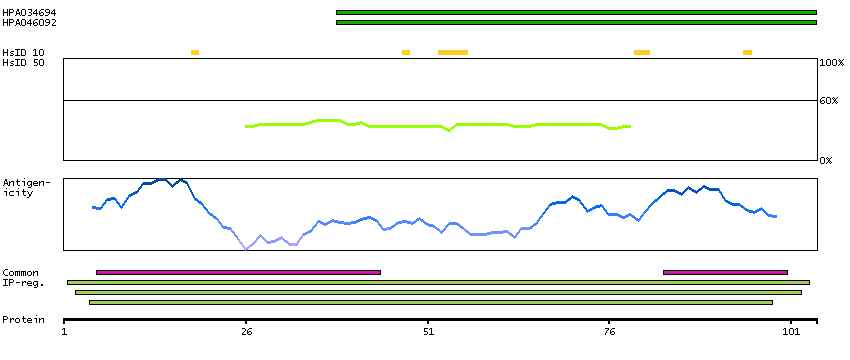
This gene encodes the fourth largest subunit of RNA polymerase II, the polymerase responsible for synthesizing messenger RNA in eukaryotes. In yeast, this polymerase subunit is associated with the polymerase under suboptimal growth conditions and may have a stress protective role. A sequence for a ribosomal pseudogene is contained within the 3' untranslated region of the transcript from this gene. [provided by RefSeq, Jul 2008]
Predicted intracellular proteins Plasma proteins RNA polymerase related proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000166 [nucleotide binding] GO:0000288 [nuclear-transcribed mRNA catabolic process, deadenylation-dependent decay] GO:0000398 [mRNA splicing, via spliceosome] GO:0000932 [cytoplasmic mRNA processing body] GO:0003697 [single-stranded DNA binding] GO:0003727 [single-stranded RNA binding] GO:0003824 [catalytic activity] GO:0003899 [DNA-directed RNA polymerase activity] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005665 [DNA-directed RNA polymerase II, core complex] GO:0006281 [DNA repair] GO:0006283 [transcription-coupled nucleotide-excision repair] GO:0006289 [nucleotide-excision repair] GO:0006351 [transcription, DNA-templated] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006367 [transcription initiation from RNA polymerase II promoter] GO:0006368 [transcription elongation from RNA polymerase II promoter] GO:0006370 [7-methylguanosine mRNA capping] GO:0008380 [RNA splicing] GO:0010467 [gene expression] GO:0016032 [viral process] GO:0031047 [gene silencing by RNA] GO:0031369 [translation initiation factor binding] GO:0031990 [mRNA export from nucleus in response to heat stress] GO:0034402 [recruitment of 3'-end processing factors to RNA polymerase II holoenzyme complex] GO:0034587 [piRNA metabolic process] GO:0035019 [somatic stem cell population maintenance] GO:0044237 [cellular metabolic process] GO:0045948 [positive regulation of translational initiation] GO:0050434 [positive regulation of viral transcription]