We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
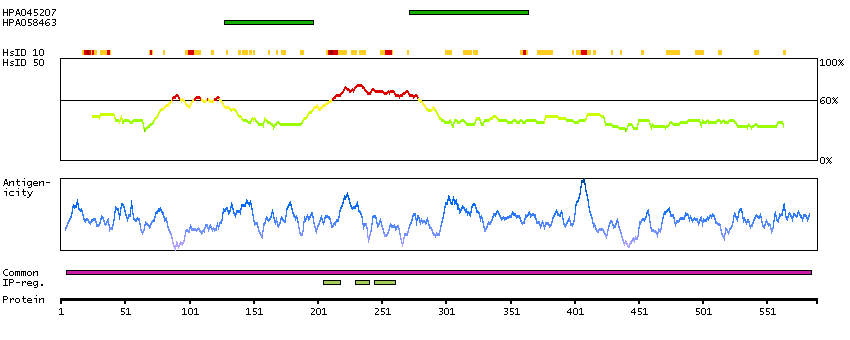
Cysteine-serine-rich nuclear protein 1 (HGNC Symbol)
Entrez gene summary
This gene encodes a protein that localizes to the nucleus and expression of this gene is induced in response to elevated levels of axin. The Wnt signalling pathway, which is negatively regulated by axin, is important in axis formation in early development and impaired regulation of this signalling pathway is often involved in tumors. A decreased level of expression of this gene in tumors compared to the level of expression in their corresponding normal tissues suggests that this gene product has a tumor suppressor function. [provided by RefSeq, Jul 2008]