We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
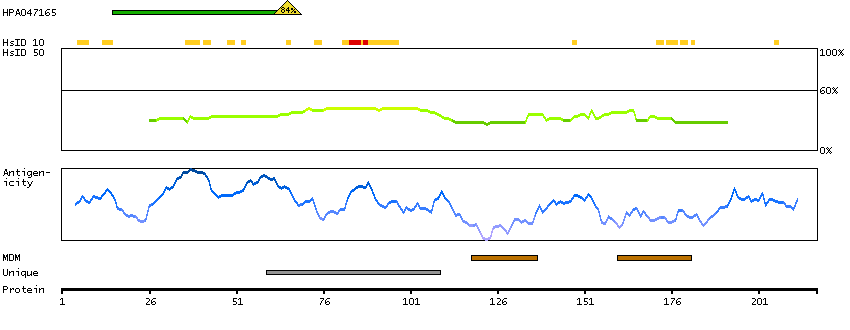
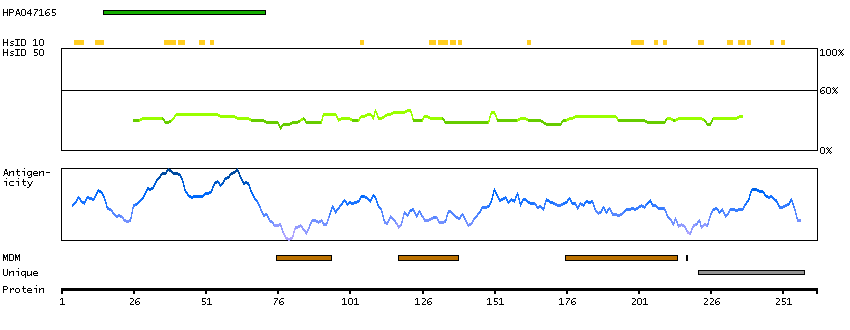
This gene encodes a putative tumor suppressor and has higher expression in p53-expressing cells than in control cells and is an immediate-early induction target of p53-mediated apoptosis. The encoded protein may suppress cell growth by inducing apoptotic cell death through the caspase 9 and mitochondrial pathways. This gene is located on human chromosome 11q24, a region frequently altered in cancers. Alternative splicing results in multiple transcript variants. Pseudogenes of this gene have been defined on chromosomes 1, 3, 7, and 8. [provided by RefSeq, Feb 2014]