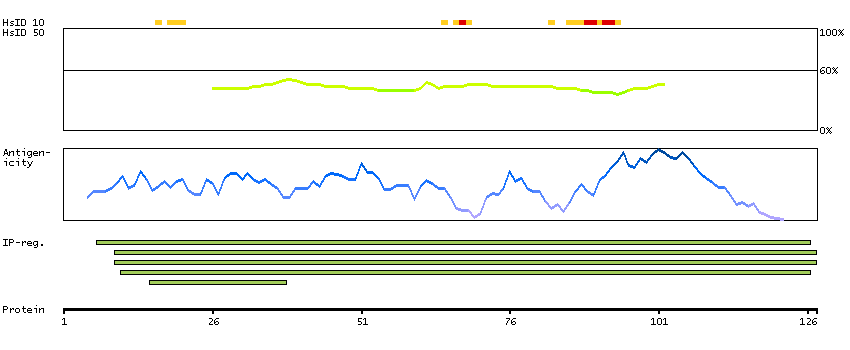
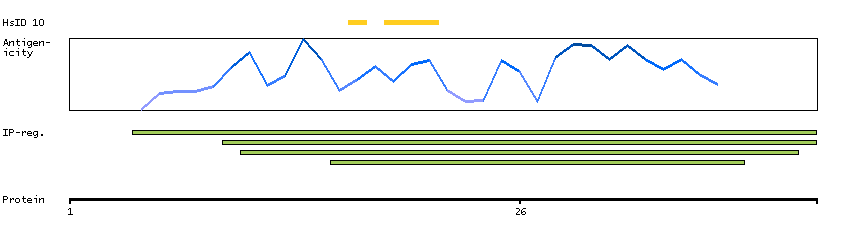
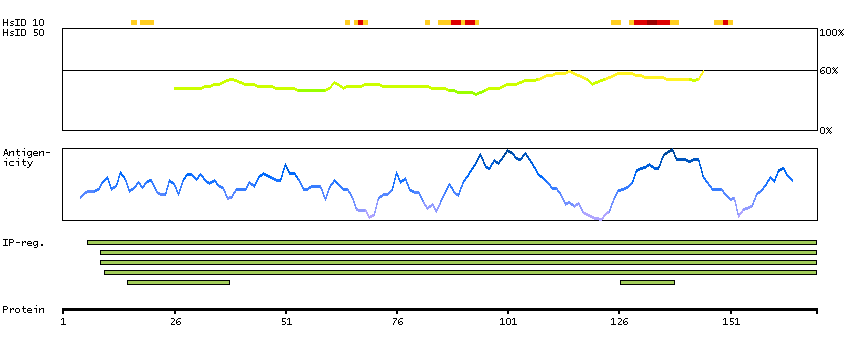
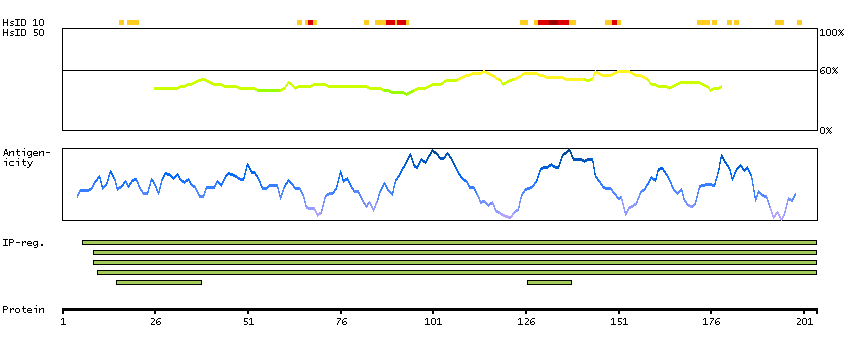
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
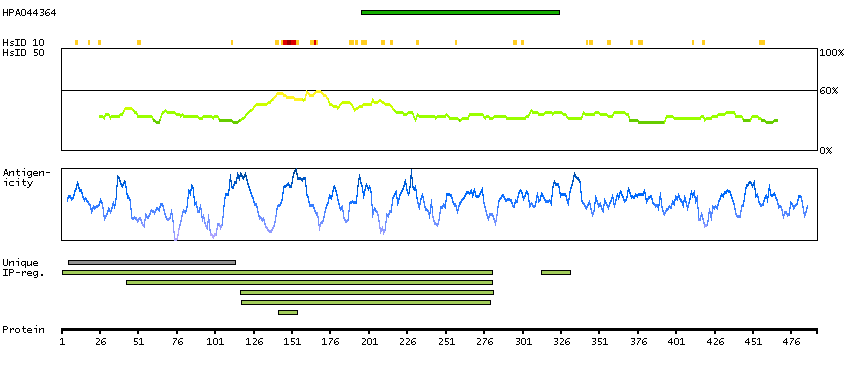
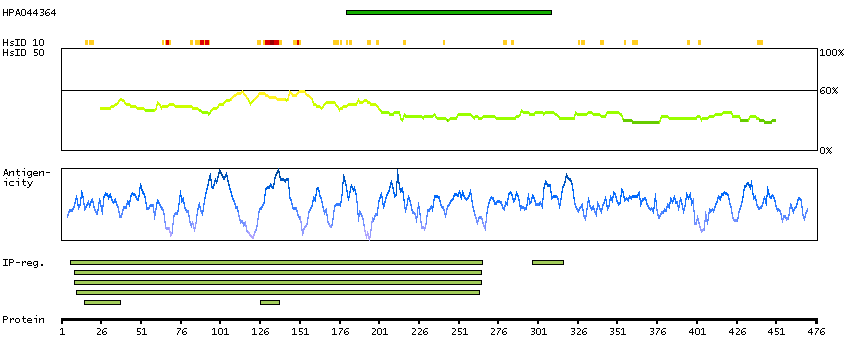
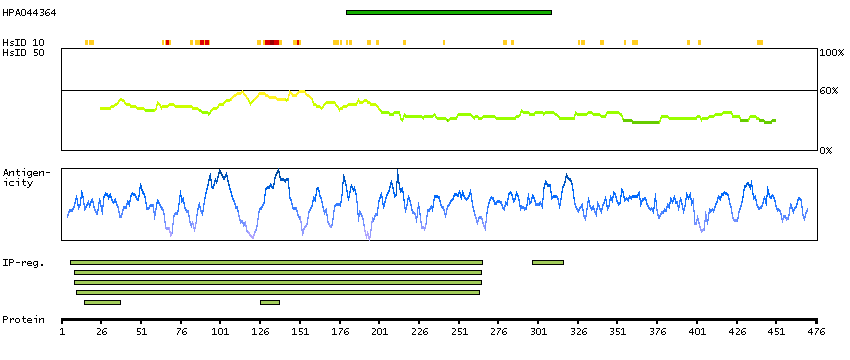
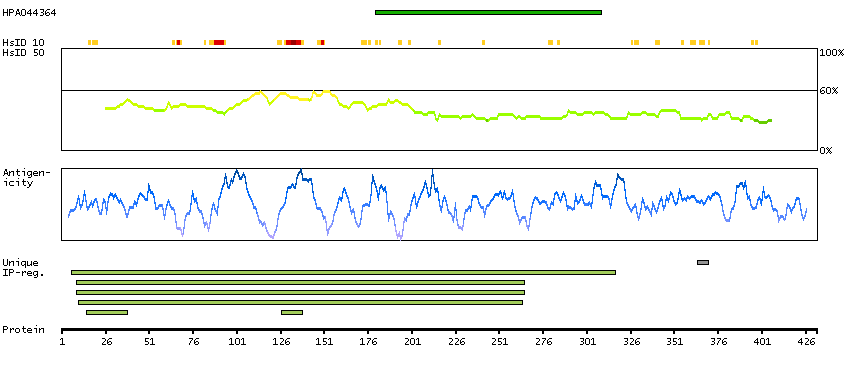
The protein encoded by this gene belongs to the Ser/Thr protein kinase family. It is required for checkpoint mediated cell cycle arrest in response to DNA damage or the presence of unreplicated DNA. This protein acts to integrate signals from ATM and ATR, two cell cycle proteins involved in DNA damage responses, that also associate with chromatin in meiotic prophase I. Phosphorylation of CDC25A protein phosphatase by this protein is required for cells to delay cell cycle progression in response to double-strand DNA breaks. Several alternatively spliced transcript variants have been found for this gene. [provided by RefSeq, Oct 2011]