We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
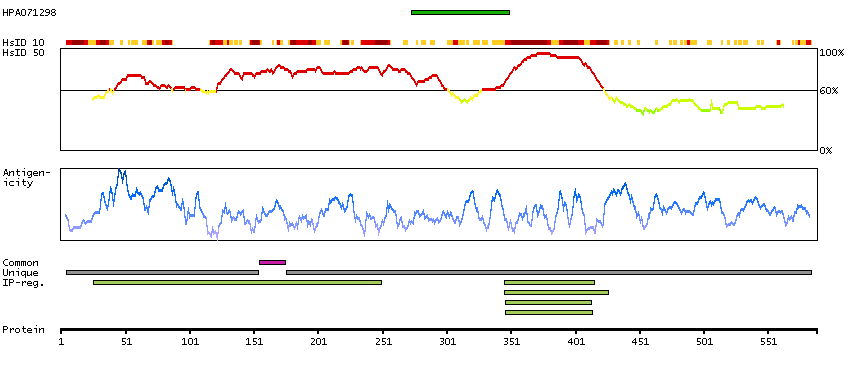
This gene encodes a member of the T cell factor/lymphoid enhancer factor family of transcription factors. These transcription factors are activated by beta catenin, mediate the Wnt signaling pathway and are antagonized by the transforming growth factor beta signaling pathway. The encoded protein contains a high mobility group-box DNA binding domain and participates in the regulation of cell cycle genes and cellular senescence. [provided by RefSeq, Nov 2010]