We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
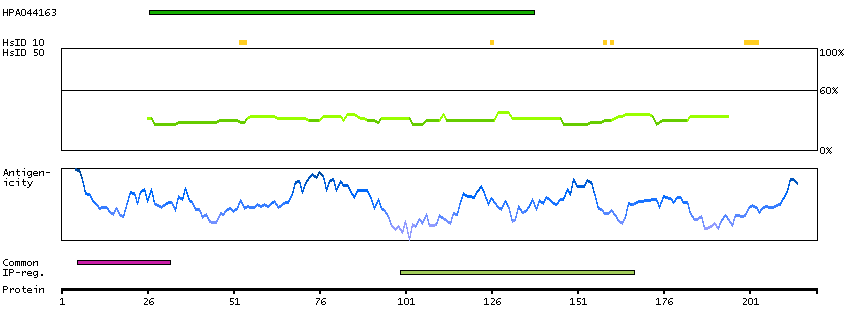
Autophagy is a process for the bulk degradation of cytosolic compartments by lysosomes. ATG10 is an E2-like enzyme involved in 2 ubiquitin-like modifications essential for autophagosome formation: ATG12 (MIM 609608)-ATG5 (MIM 604261) conjugation and modification of a soluble form of MAP-LC3 (MAP1LC3A; MIM 601242), a homolog of yeast Apg8, to a membrane-bound form (Nemoto et al., 2003 [PubMed 12890687]).[supplied by OMIM, Mar 2008]