We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
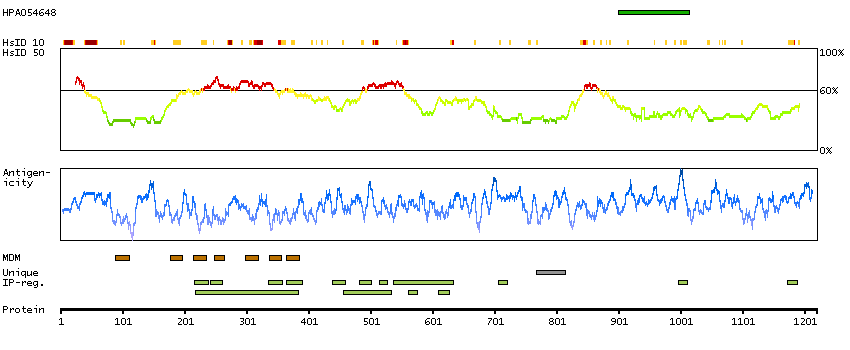
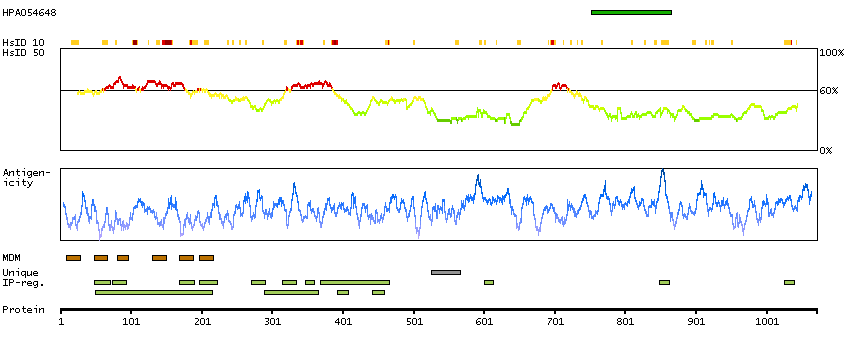
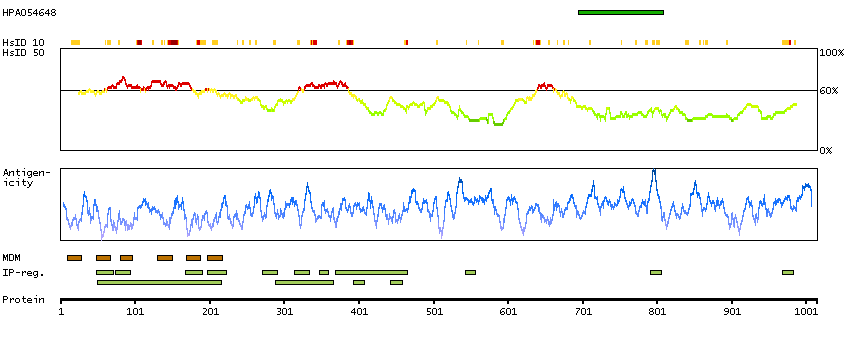
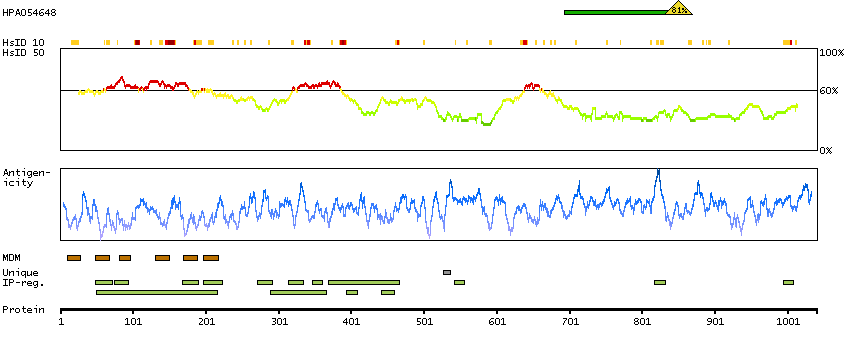
Potassium channel, calcium activated large conductance subfamily M alpha, member 1 (HGNC Symbol)
Entrez gene summary
MaxiK channels are large conductance, voltage and calcium-sensitive potassium channels which are fundamental to the control of smooth muscle tone and neuronal excitability. MaxiK channels can be formed by 2 subunits: the pore-forming alpha subunit, which is the product of this gene, and the modulatory beta subunit. Intracellular calcium regulates the physical association between the alpha and beta subunits. Alternatively spliced transcript variants encoding different isoforms have been identified. [provided by RefSeq, Jul 2008]