We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
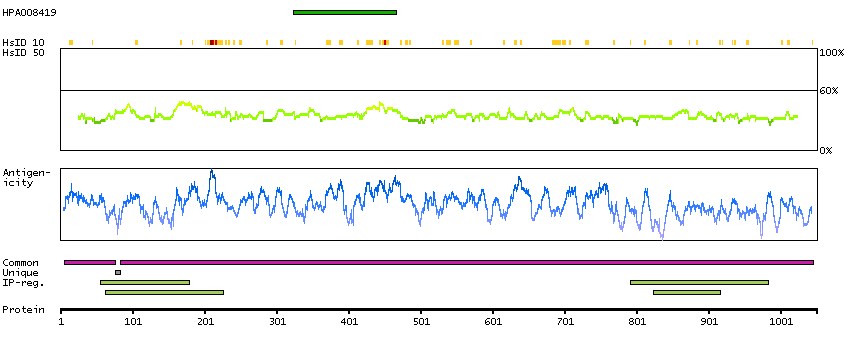
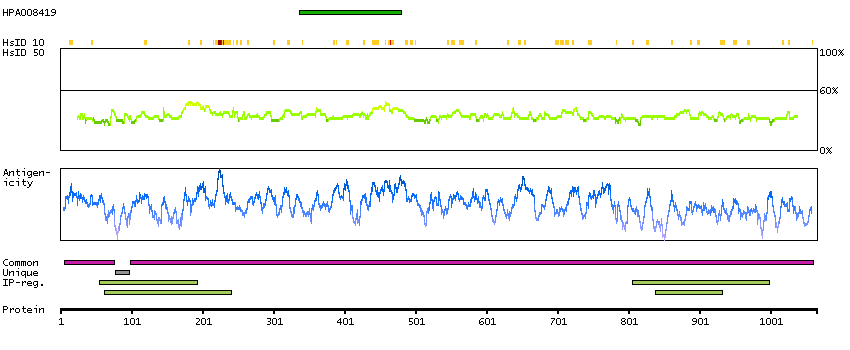
BUB1 mitotic checkpoint serine/threonine kinase B (HGNC Symbol)
Entrez gene summary
This gene encodes a kinase involved in spindle checkpoint function. The protein has been localized to the kinetochore and plays a role in the inhibition of the anaphase-promoting complex/cyclosome (APC/C), delaying the onset of anaphase and ensuring proper chromosome segregation. Impaired spindle checkpoint function has been found in many forms of cancer. [provided by RefSeq, Jul 2008]