We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Fanconi anemia, complementation group C (HGNC Symbol)
Entrez gene summary
The Fanconi anemia complementation group (FANC) currently includes FANCA, FANCB, FANCC, FANCD1 (also called BRCA2), FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ (also called BRIP1), FANCL, FANCM and FANCN (also called PALB2). The previously defined group FANCH is the same as FANCA. Fanconi anemia is a genetically heterogeneous recessive disorder characterized by cytogenetic instability, hypersensitivity to DNA crosslinking agents, increased chromosomal breakage, and defective DNA repair. The members of the Fanconi anemia complementation group do not share sequence similarity; they are related by their assembly into a common nuclear protein complex. This gene encodes the protein for complementation group C. [provided by RefSeq, Jul 2008]
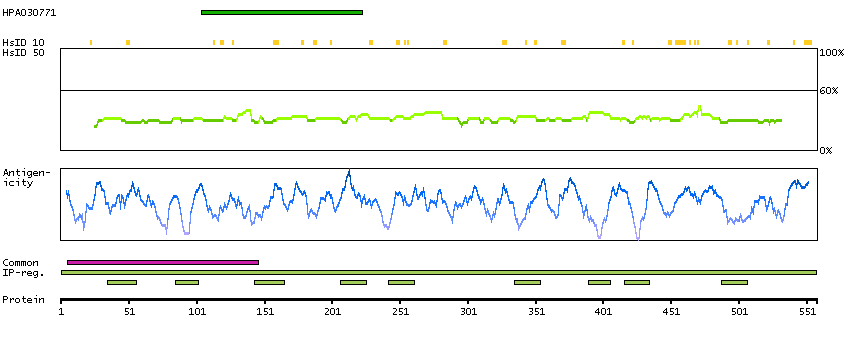
Q00597 [Direct mapping] Fanconi anemia group C protein A0A024R9N2 [Target identity:100%; Query identity:100%] Fanconi anemia, complementation group C, isoform CRA_a
Show all
THUMBUP predicted membrane proteins Predicted intracellular proteins Plasma proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Splicing Mutations COSMIC Nonsense Mutations COSMIC Missense Mutations COSMIC Large Deletions COSMIC Germline Mutations COSMIC Frameshift Mutations Disease related genes Protein evidence (Ezkurdia et al 2014)
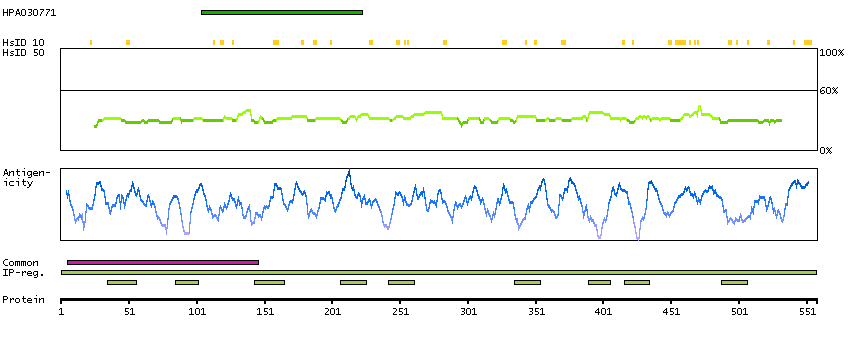
Q00597 [Direct mapping] Fanconi anemia group C protein A0A024R9N2 [Target identity:100%; Query identity:100%] Fanconi anemia, complementation group C, isoform CRA_a
Show all
THUMBUP predicted membrane proteins Predicted intracellular proteins Plasma proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Splicing Mutations COSMIC Nonsense Mutations COSMIC Missense Mutations COSMIC Large Deletions COSMIC Germline Mutations COSMIC Frameshift Mutations Disease related genes Protein evidence (Ezkurdia et al 2014)