We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
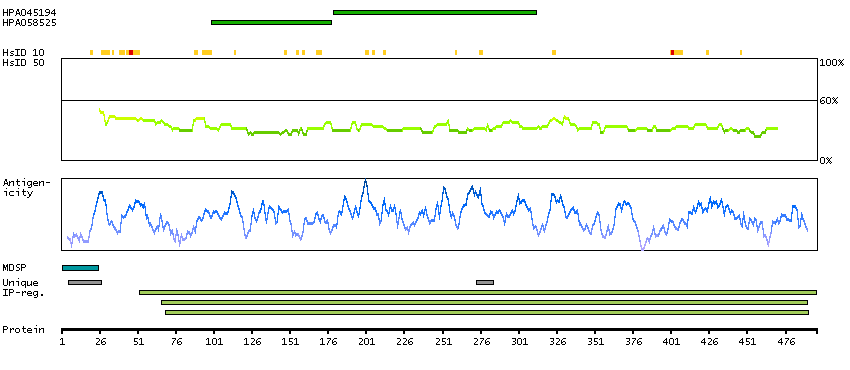
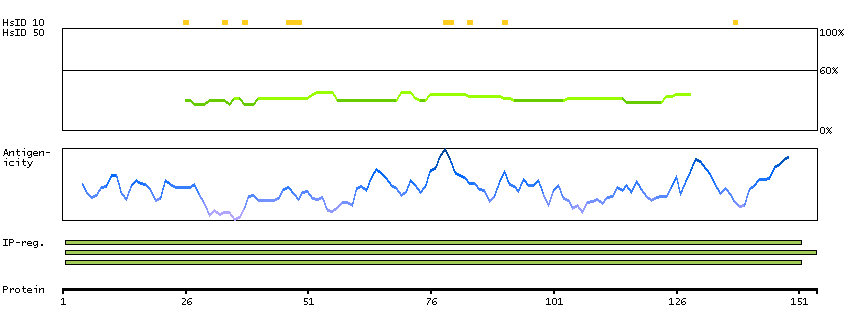
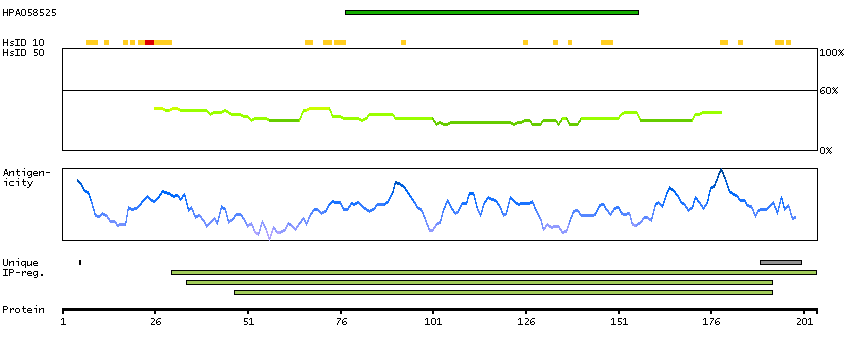
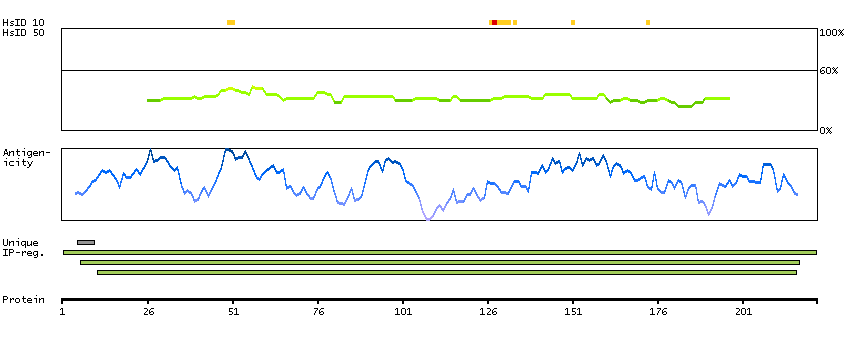
ADPGK (EC 2.7.1.147) catalyzes the ADP-dependent phosphorylation of glucose to glucose-6-phosphate and may play a role in glycolysis, possibly during ischemic conditions (Ronimus and Morgan, 2004 [PubMed 14975750]).[supplied by OMIM, Mar 2008]