We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
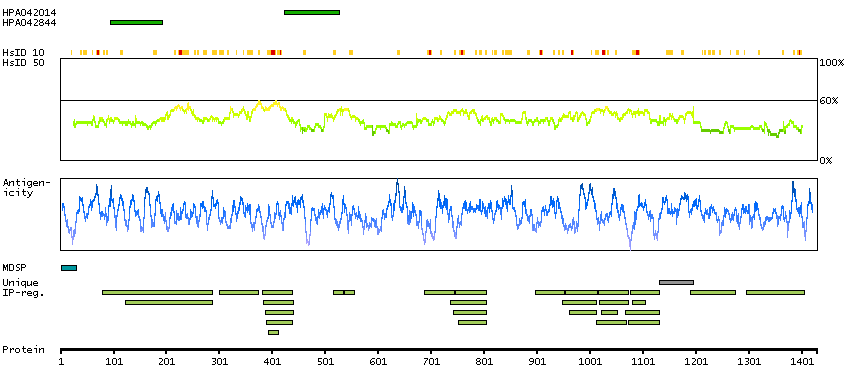
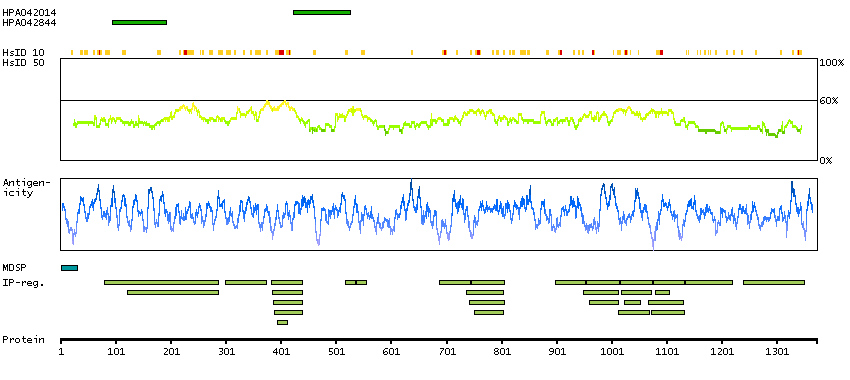
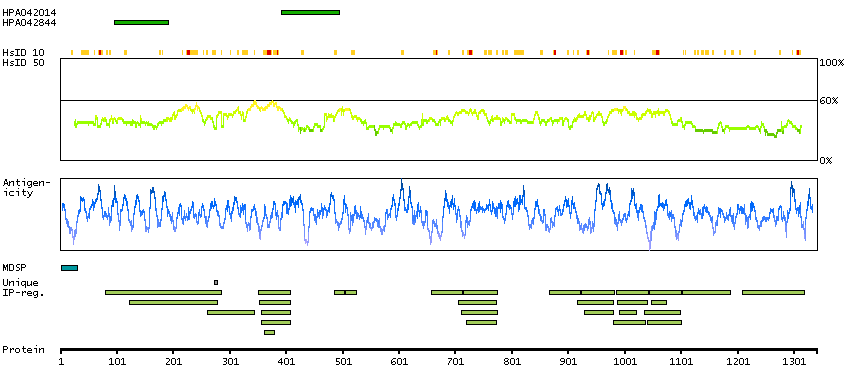
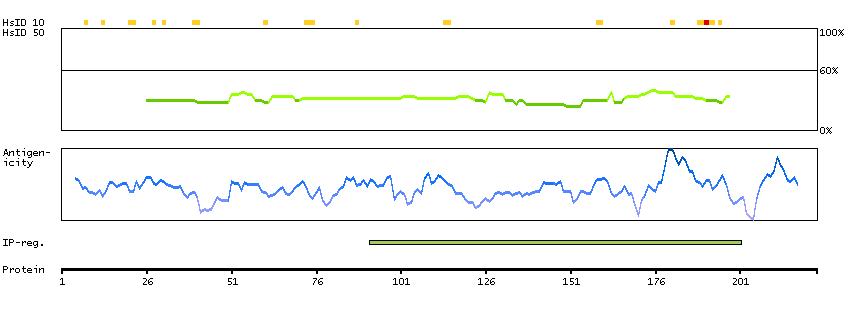
ADAM metallopeptidase with thrombospondin type 1 motif, 13 (HGNC Symbol)
Entrez gene summary
This gene encodes a member of a family of proteins containing several distinct regions, including a metalloproteinase domain, a disintegrin-like domain, and a thrombospondin type 1 (TS) motif. The enzyme encoded by this gene specifically cleaves von Willebrand Factor (vWF). Defects in this gene are associated with thrombotic thrombocytopenic purpura. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jul 2013]