We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
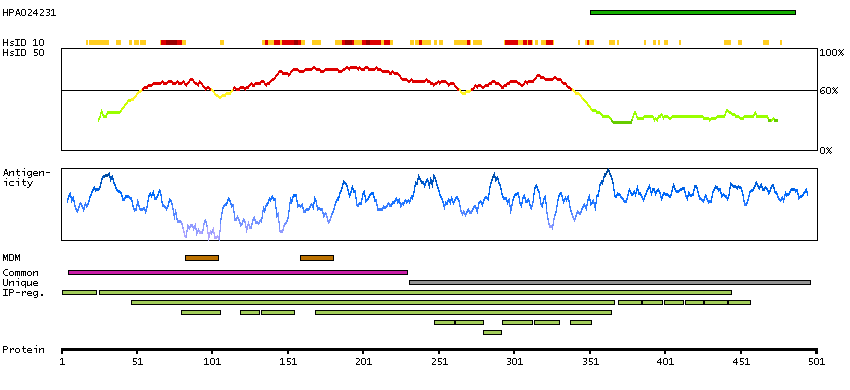
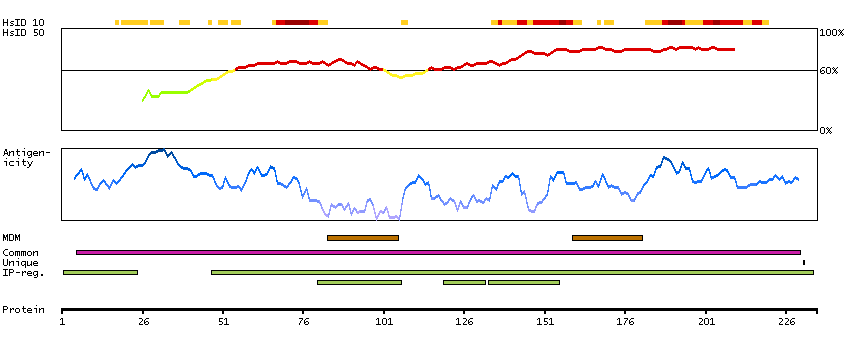
Potassium channel, inwardly rectifying subfamily J, member 3 (HGNC Symbol)
Entrez gene summary
Potassium channels are present in most mammalian cells, where they participate in a wide range of physiologic responses. The protein encoded by this gene is an integral membrane protein and inward-rectifier type potassium channel. The encoded protein, which has a greater tendency to allow potassium to flow into a cell rather than out of a cell, is controlled by G-proteins and plays an important role in regulating heartbeat. It associates with three other G-protein-activated potassium channels to form a heteromultimeric pore-forming complex that also couples to neurotransmitter receptors in the brain and whereby channel activation can inhibit action potential firing by hyperpolarizing the plasma membrane. These multimeric G-protein-gated inwardly-rectifying potassium (GIRK) channels may play a role in the pathophysiology of epilepsy, addiction, Down's syndrome, ataxia, and Parkinson's disease. Alternative splicing results in multiple transcript variants encoding distinct proteins. [provided by RefSeq, May 2012]