We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
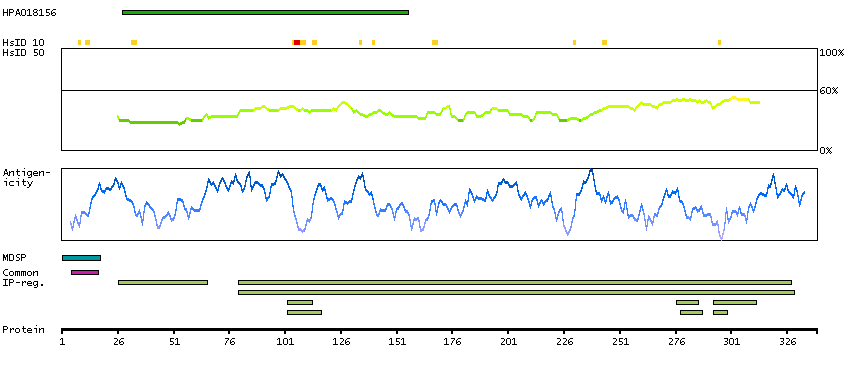
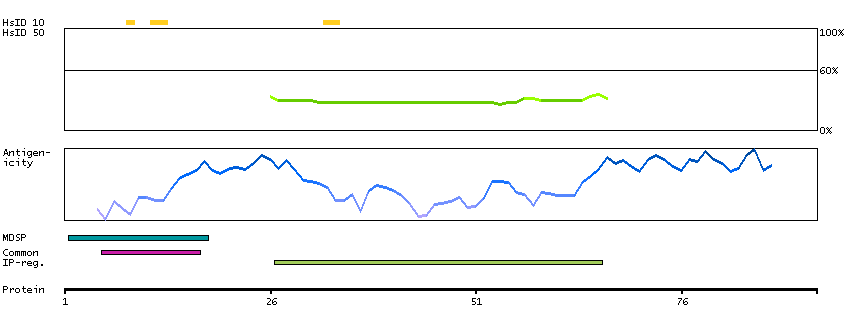
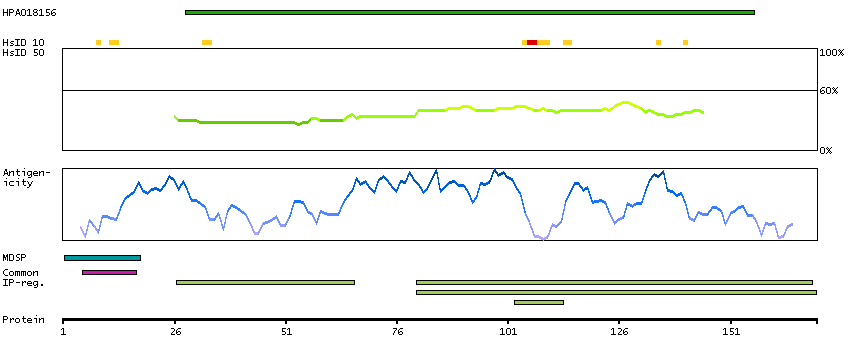
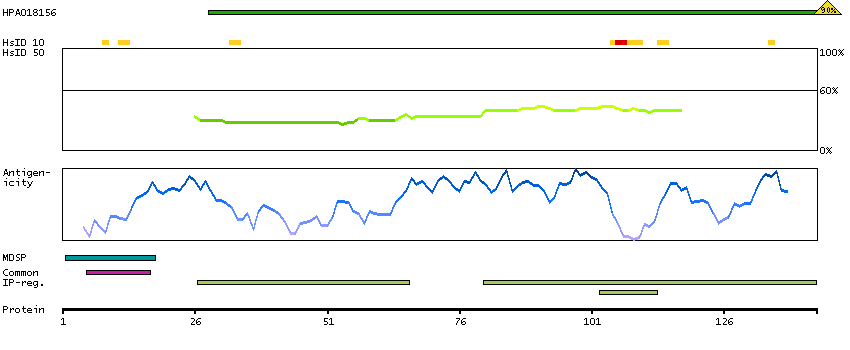
This gene encodes a member of the C1 family of peptidases. Alternative splicing of this gene results in multiple transcript variants. At least one of these variants encodes a preproprotein that is proteolytically processed to generate multiple protein products. These products include the cathepsin B light and heavy chains, which can dimerize to form the double chain form of the enzyme. This enzyme is a lysosomal cysteine protease with both endopeptidase and exopeptidase activity that may play a role in protein turnover. It is also known as amyloid precursor protein secretase and is involved in the proteolytic processing of amyloid precursor protein (APP). Incomplete proteolytic processing of APP has been suggested to be a causative factor in Alzheimer's disease, the most common cause of dementia. Overexpression of the encoded protein has been associated with esophageal adenocarcinoma and other tumors. Multiple pseudogenes of this gene have been identified. [provided by RefSeq, Nov 2015]