We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
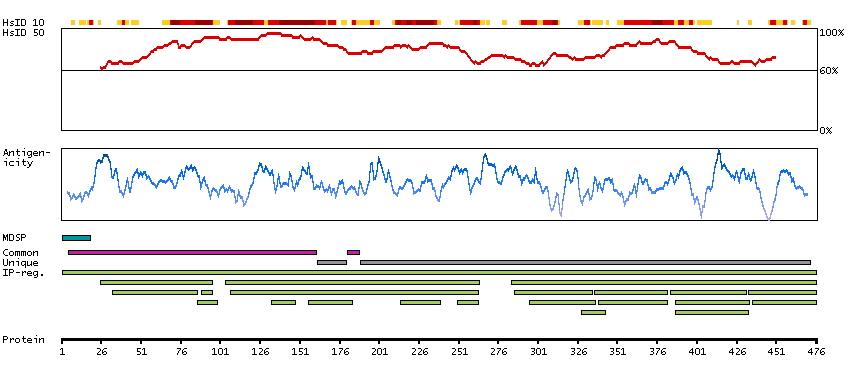
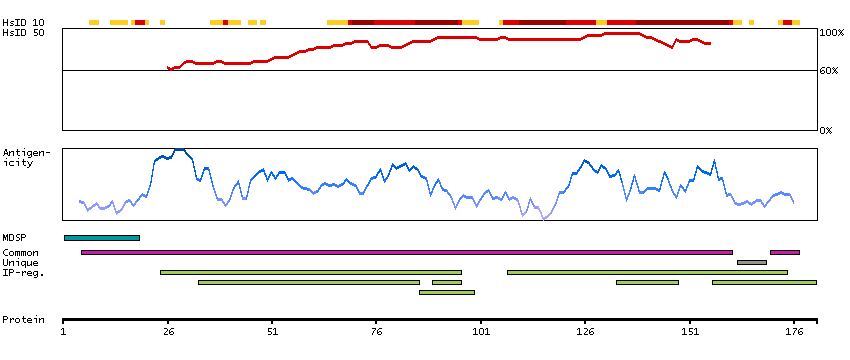
Proteins of the matrix metalloproteinase (MMP) family are involved in the breakdown of extracellular matrix in normal physiological processes, such as embryonic development, reproduction, and tissue remodeling, as well as in disease processes, such as arthritis and metastasis. Most MMP's are secreted as inactive proproteins which are activated when cleaved by extracellular proteinases. The enzyme encoded by this gene degrades proteoglycans and fibronectin. The gene is part of a cluster of MMP genes which localize to chromosome 11q22.3. [provided by RefSeq, Jul 2008]
Enzymes ENZYME proteins Hydrolases Peptidases Metallopeptidases Predicted secreted proteins Secreted proteins predicted by MDSEC SignalP predicted secreted proteins Phobius predicted secreted proteins SPOCTOPUS predicted secreted proteins Cancer-related genes Candidate cancer biomarkers FDA approved drug targets Small molecule drugs Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)