We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
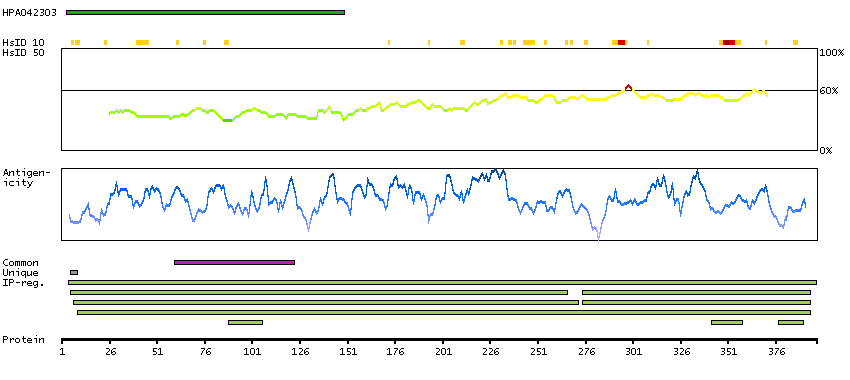
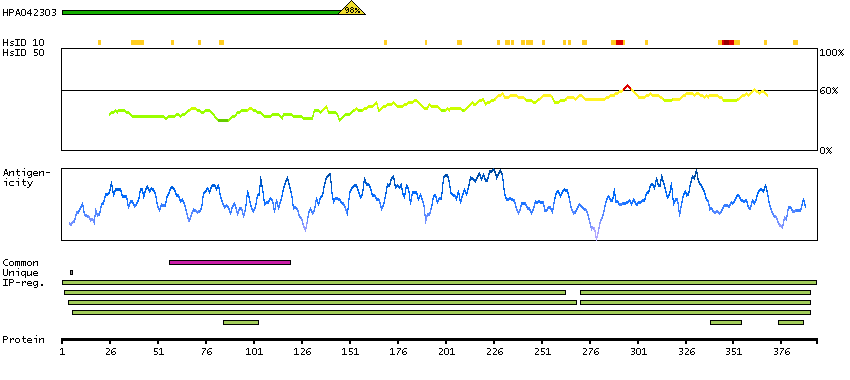
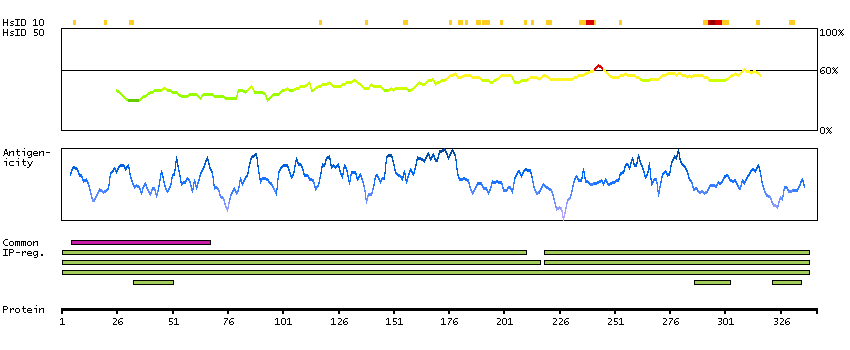
The encoded protein catalyzes the last step of the mitochondrial fatty acid beta-oxidation spiral. Unlike most mitochondrial matrix proteins, it contains a non-cleavable amino-terminal targeting signal. [provided by RefSeq, Jul 2008]
Enzymes ENZYME proteins Transferases SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003824 [catalytic activity] GO:0003988 [acetyl-CoA C-acyltransferase activity] GO:0005515 [protein binding] GO:0005739 [mitochondrion] GO:0005743 [mitochondrial inner membrane] GO:0006631 [fatty acid metabolic process] GO:0006695 [cholesterol biosynthetic process] GO:0008152 [metabolic process] GO:0016747 [transferase activity, transferring acyl groups other than amino-acyl groups] GO:0044822 [poly(A) RNA binding] GO:0070062 [extracellular exosome] GO:0071456 [cellular response to hypoxia] GO:1901029 [negative regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway] GO:1902109 [negative regulation of mitochondrial membrane permeability involved in apoptotic process]