We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Expression in basal membrane in photoreceptor layer in retina.
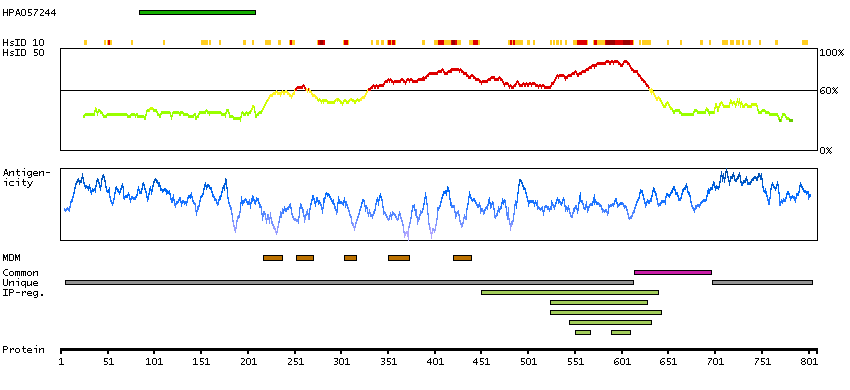
DATA RELIABILITY
Data reliability description
Presumed off target binding observed and disregarded. External characterization data supports antibody staining but no internal RNA data available for correlation.
This gene encodes the beta subunit of a cyclic nucleotide-gated ion channel. The encoded beta subunit appears to play a role in modulation of channel function in cone photoreceptors. This heterotetrameric channel is necessary for sensory transduction, and mutations in this gene have been associated with achromatopsia 3, progressive cone dystrophy, and juvenile macular degeneration, also known as Stargardt Disease. [provided by RefSeq, Feb 2010]