We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
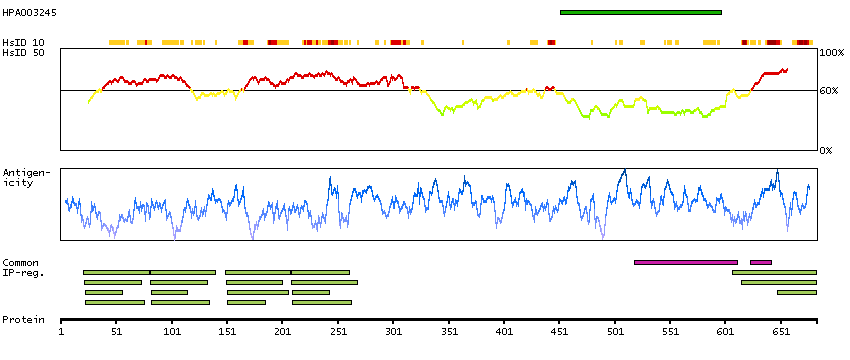
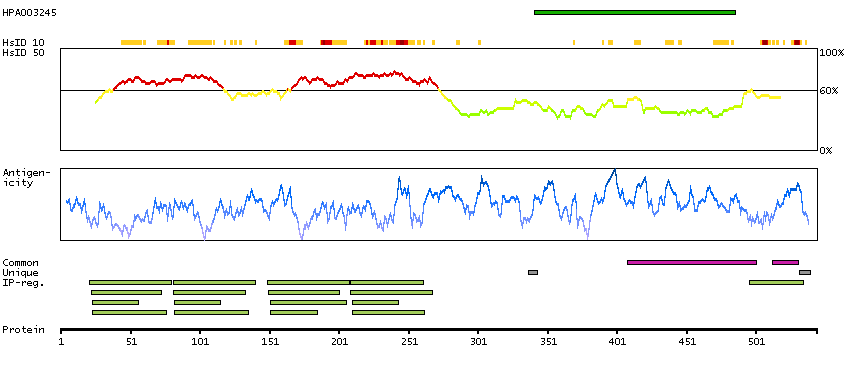
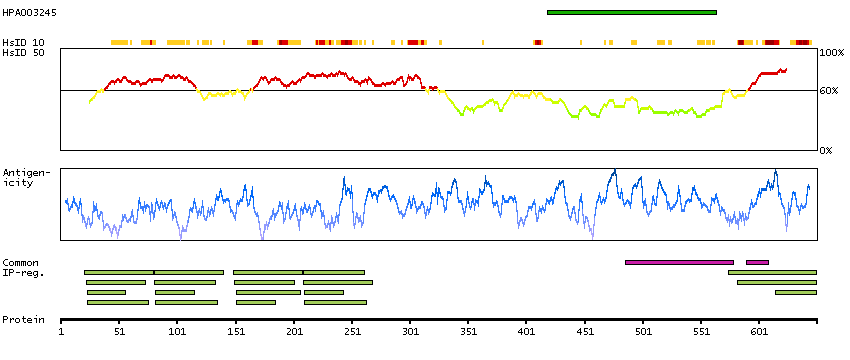
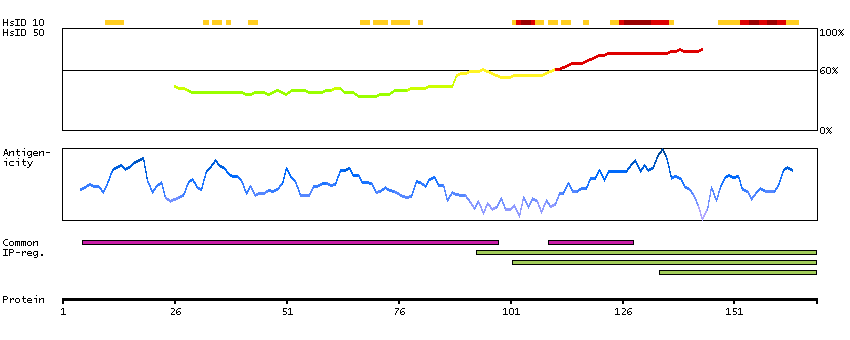
Actin binding LIM protein family, member 3 (HGNC Symbol)
Entrez gene summary
The LIM domain is a double zinc finger structure that promotes protein-protein interactions. LIM domain proteins, such as ABLIM3, play roles in embryonic development, cell lineage determination, and cancer (Krupp et al., 2006 [PubMed 16328021]).[supplied by OMIM, Mar 2008]