We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
RNA tissue category: Group enriched (appendix, duodenum, small intestine, spleen)
GTEx dataset
Organ
Expression
Alphabetical
RNA tissue category: Group enriched (small intestine, spleen)
FANTOM5 dataset
Organ
Expression
Alphabetical
RNA tissue category: Group enriched (colon, small intestine)
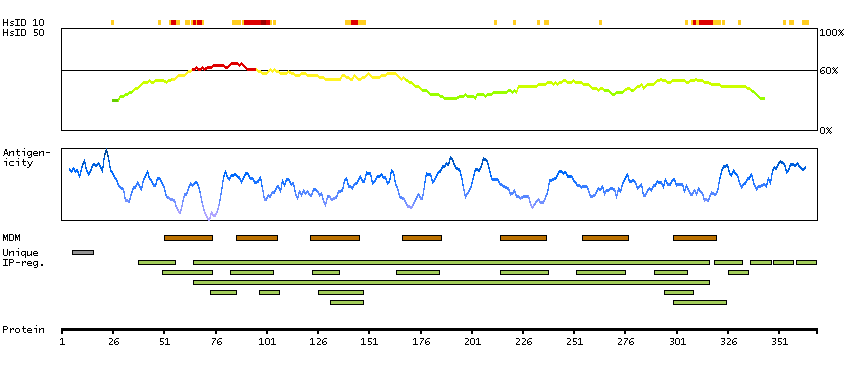
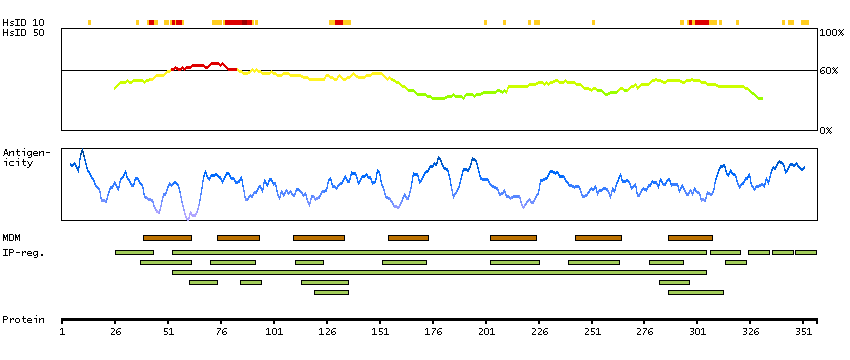
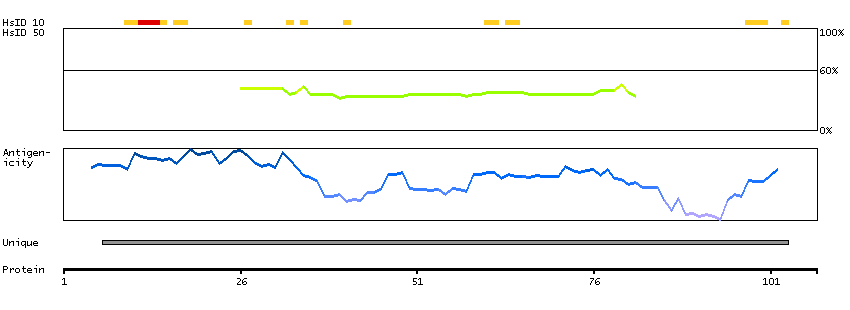
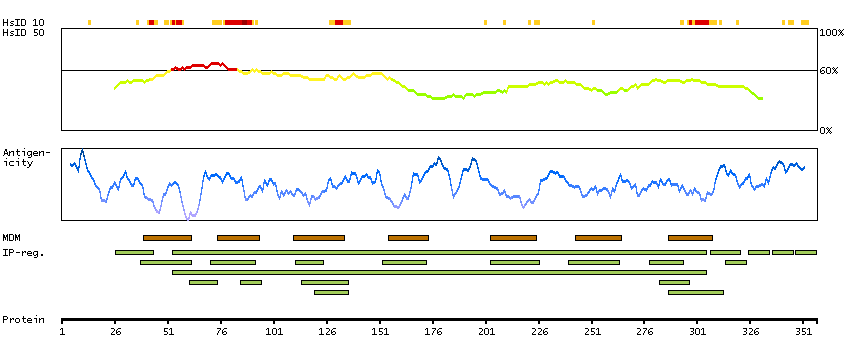
GENE INFORMATION
Gene name
CCR9 (HGNC Symbol)
Synonyms
CDw199, GPR-9-6, GPR28
Description
Chemokine (C-C motif) receptor 9 (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene is a member of the beta chemokine receptor family. It is predicted to be a seven transmembrane protein similar to G protein-coupled receptors. Chemokines and their receptors are key regulators of the thymocytes migration and maturation in normal and inflammation conditions. The specific ligand of this receptor is CCL25. It has been found that this gene is differentially expressed by T lymphocytes of small intestine and colon, suggested a role in the thymocytes recruitment and development that may permit functional specialization of immune responses in different segment of the gastrointestinal tract. This gene is mapped to the chemokine receptor gene cluster region. Two alternatively spliced transcript variants have been described. Multiple transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jan 2012]