We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
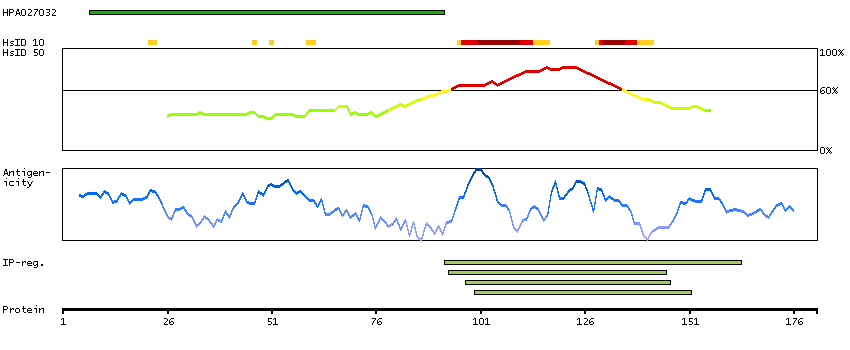
Achaete-scute family bHLH transcription factor 3 (HGNC Symbol)
Entrez gene summary
Basic helix-loop-helix transcription factors, such as ASCL3, are essential for the determination of cell fate and the development and differentiation of numerous tissues (Jonsson et al., 2004 [PubMed 15475265]).[supplied by OMIM, Mar 2008]