We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
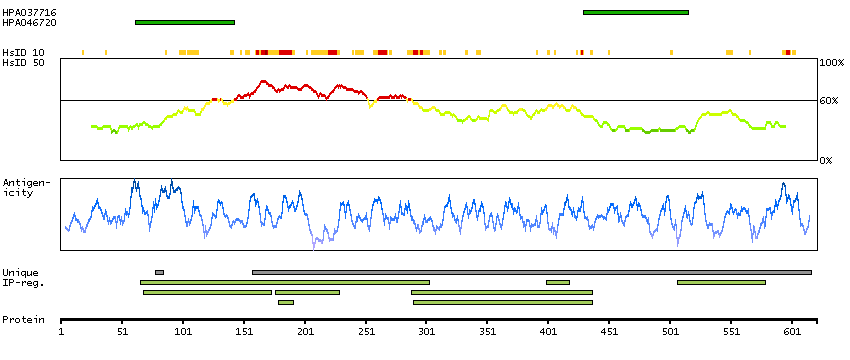
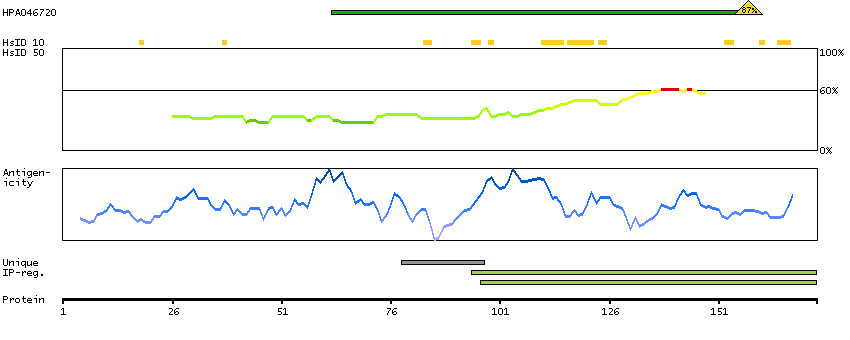
Acyl-CoA dehydrogenase family, member 9 (HGNC Symbol)
Entrez gene summary
This gene encodes a member of the acyl-CoA dehydrogenase family. Members of this family of proteins localize to the mitochondria and catalyze the rate-limiting step in the beta-oxidation of fatty acyl-CoA. The encoded protein is specifically active toward palmitoyl-CoA and long-chain unsaturated substrates. Mutations in this gene cause acyl-CoA dehydrogenase family member type 9 deficiency. Alternate splicing results in multiple transcript variants.[provided by RefSeq, Mar 2010]
Q9H845 [Direct mapping] Acyl-CoA dehydrogenase family member 9, mitochondrial
Show all
SPOCTOPUS predicted membrane proteins Phobius predicted secreted proteins Predicted intracellular proteins Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000062 [fatty-acyl-CoA binding] GO:0003995 [acyl-CoA dehydrogenase activity] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005739 [mitochondrion] GO:0008152 [metabolic process] GO:0009055 [electron carrier activity] GO:0016627 [oxidoreductase activity, acting on the CH-CH group of donors] GO:0030425 [dendrite] GO:0032981 [mitochondrial respiratory chain complex I assembly] GO:0033539 [fatty acid beta-oxidation using acyl-CoA dehydrogenase] GO:0050660 [flavin adenine dinucleotide binding] GO:0052890 [oxidoreductase activity, acting on the CH-CH group of donors, with a flavin as acceptor] GO:0055088 [lipid homeostasis] GO:0055114 [oxidation-reduction process]