We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
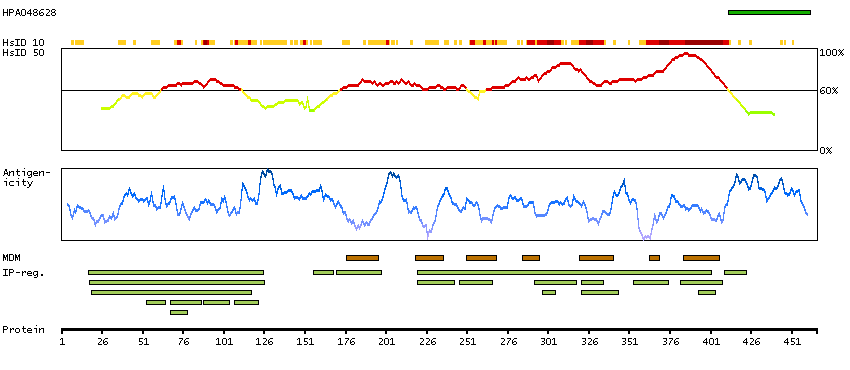
Potassium channel, voltage gated modifier subfamily G, member 2 (HGNC Symbol)
Entrez gene summary
Voltage-gated potassium (Kv) channels represent the most complex class of voltage-gated ion channels from both functional and structural standpoints. Their diverse functions include regulating neurotransmitter release, heart rate, insulin secretion, neuronal excitability, epithelial electrolyte transport, smooth muscle contraction, and cell volume. This gene encodes a member of the potassium channel, voltage-gated, subfamily G. This member is a gamma subunit of the voltage-gated potassium channel. The delayed-rectifier type channels containing this subunit may contribute to cardiac action potential repolarization. [provided by RefSeq, Jul 2008]