We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
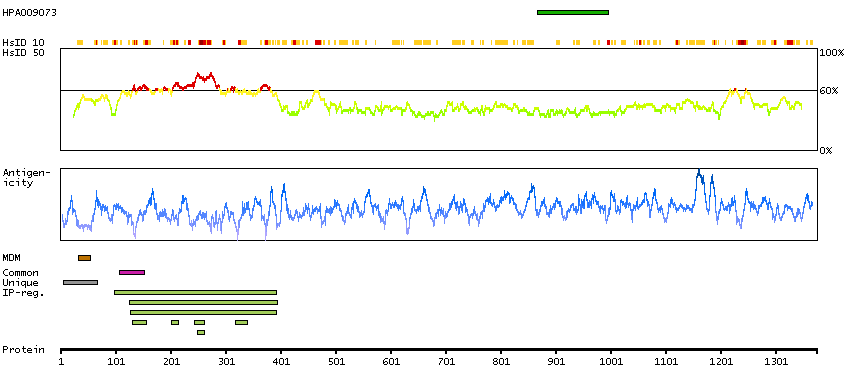
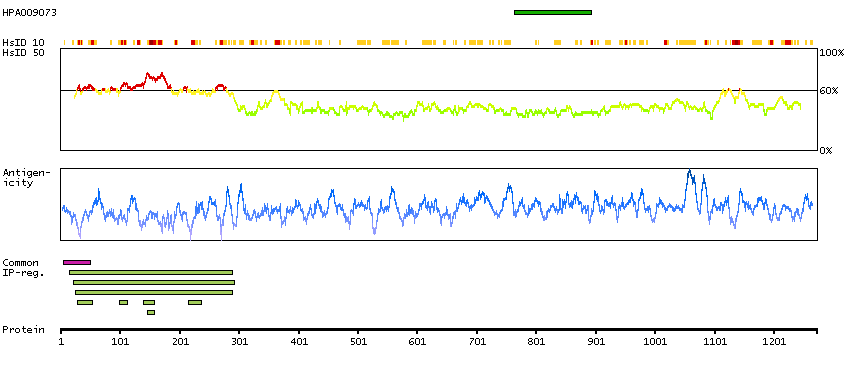
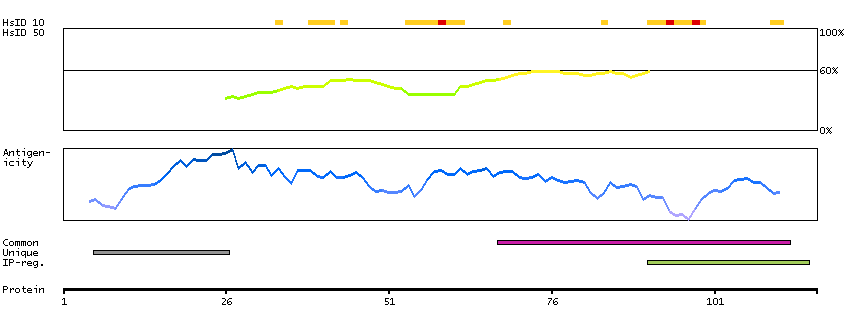
The protein encoded by this gene contains a tyrosine kinase domain at the N-terminus and a proline-rich domain at the C-terminus. This gene is induced during apoptosis, and expression of this gene may be a necessary pre-requisite for the induction of growth arrest and/or apoptosis of myeloid precursor cells. This gene has been shown to produce neuronal differentiation in a neuroblastoma cell line. Two transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jun 2011]