We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
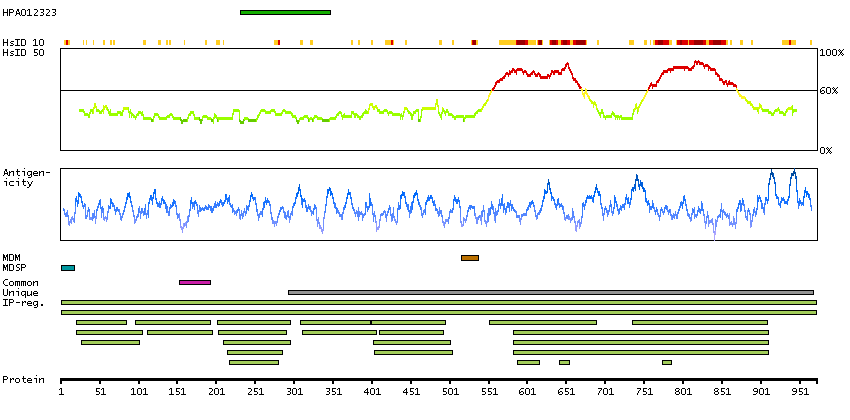
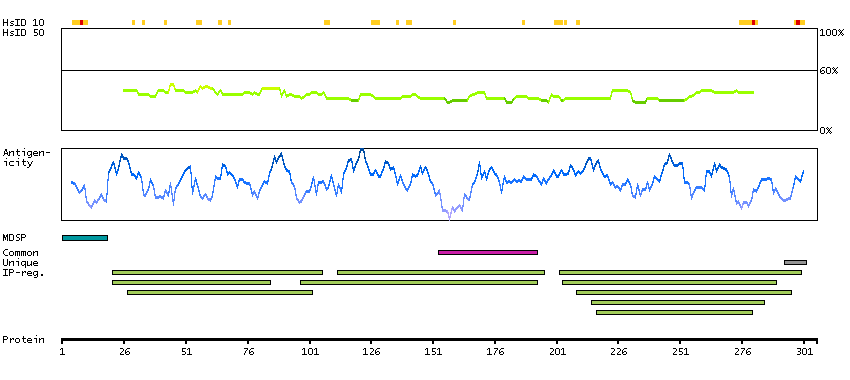
The protein encoded by this gene is the receptor for colony stimulating factor 1, a cytokine which controls the production, differentiation, and function of macrophages. This receptor mediates most if not all of the biological effects of this cytokine. Ligand binding activates the receptor kinase through a process of oligomerization and transphosphorylation. The encoded protein is a tyrosine kinase transmembrane receptor and member of the CSF1/PDGF receptor family of tyrosine-protein kinases. Mutations in this gene have been associated with a predisposition to myeloid malignancy. The first intron of this gene contains a transcriptionally inactive ribosomal protein L7 processed pseudogene oriented in the opposite direction. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Dec 2013]