We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
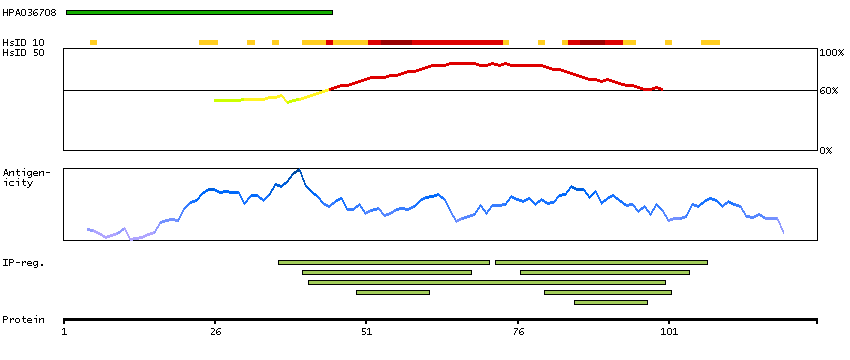
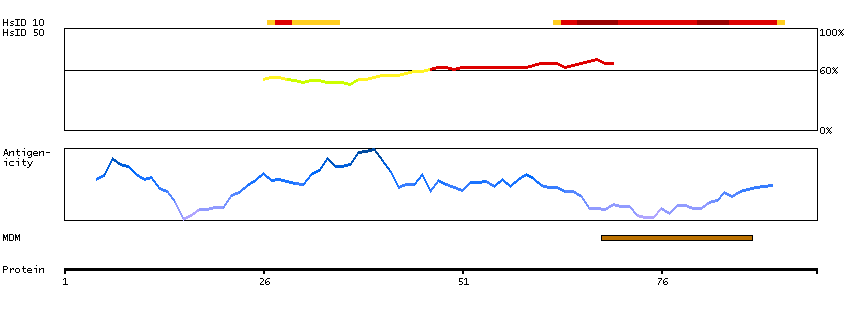
This gene encodes a protein with high similarity to the calcium-binding proteins of the calmodulin family. The encoded protein contains two EF-hand domains and potential calcium-binding sites. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jul 2008]