We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
The protein encoded by this gene upregulates trancriptional activation by the Wilms tumor protein and interacts with many other proteins, including CTNNB1, APC, AXIN1, and AXIN2. Defects in this gene are a cause of osteopathia striata with cranial sclerosis (OSCS). [provided by RefSeq, May 2010]
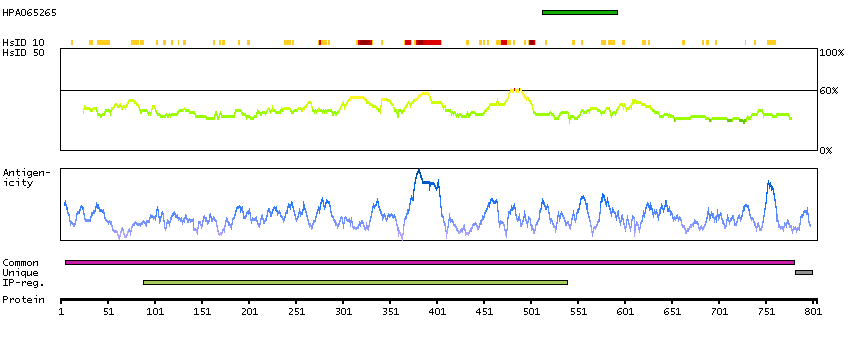
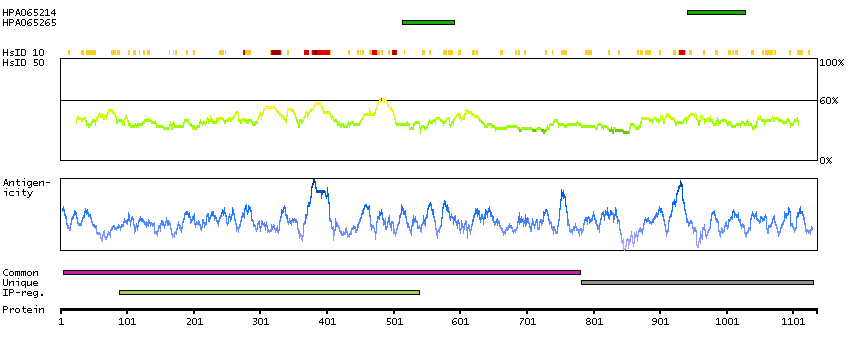
Q5JTC6 [Direct mapping] APC membrane recruitment protein 1
Show all
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Nonsense Mutations COSMIC Missense Mutations COSMIC Large Deletions COSMIC Frameshift Mutations Disease related genes Protein evidence (Kim et al 2014)