We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
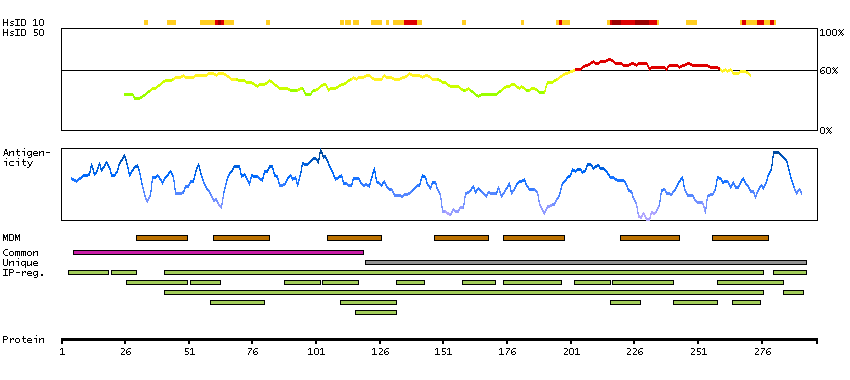
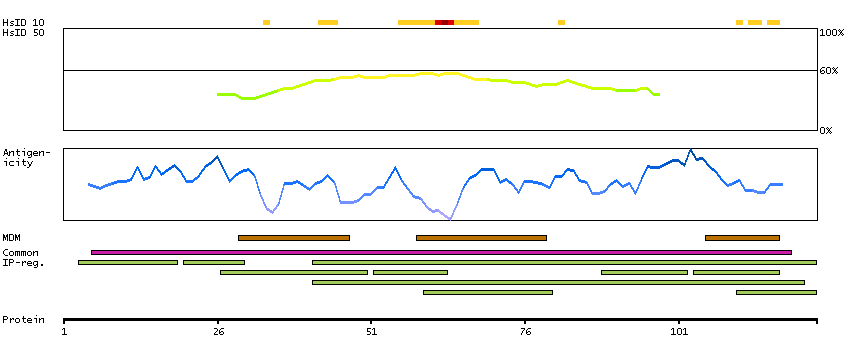
MC2R encodes one member of the five-member G-protein associated melanocortin receptor family. Melanocortins (melanocyte-stimulating hormones and adrenocorticotropic hormone) are peptides derived from pro-opiomelanocortin (POMC). MC2R is selectively activated by adrenocorticotropic hormone, whereas the other four melanocortin receptors recognize a variety of melanocortin ligands. Mutations in MC2R can result in familial glucocorticoid deficiency. Alternate transcript variants have been found for this gene. [provided by RefSeq, May 2014]