We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
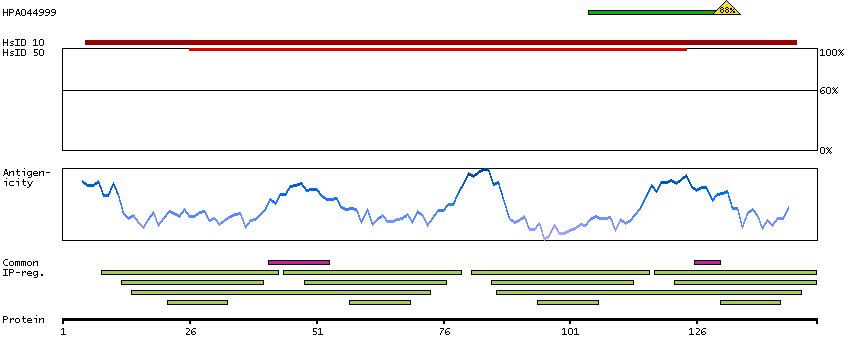
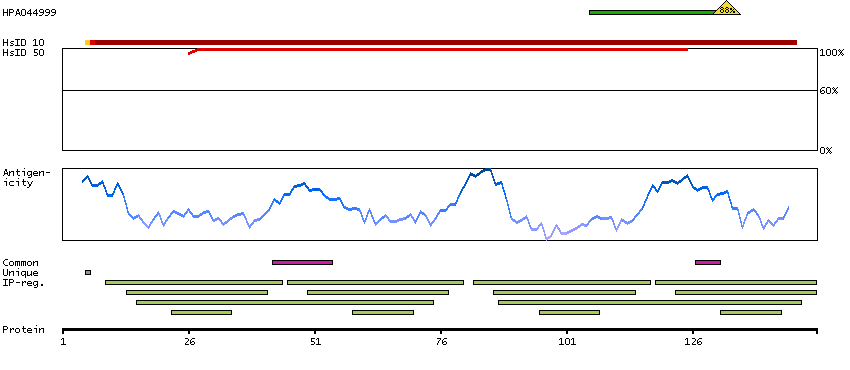
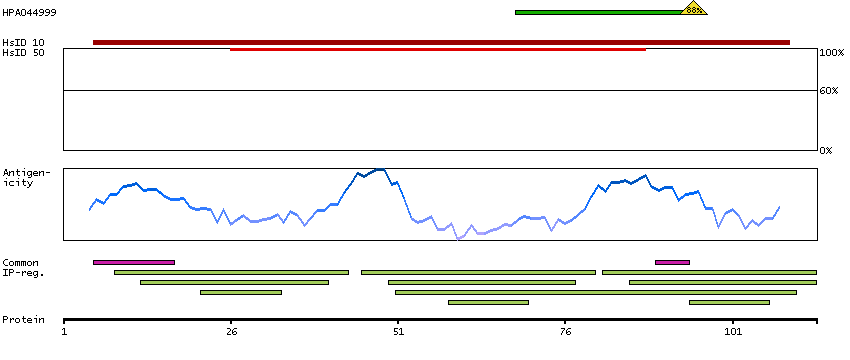
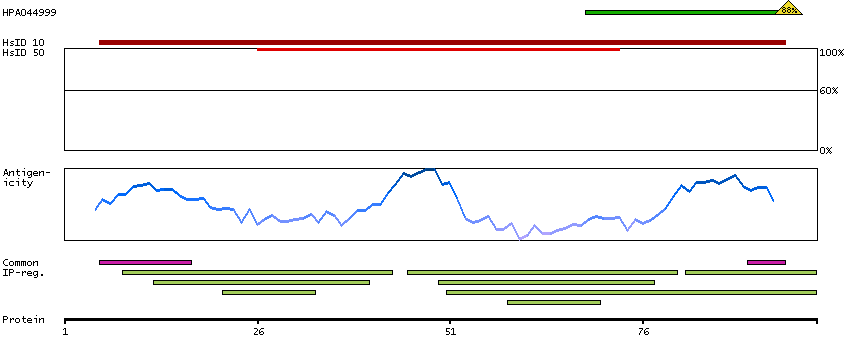
This gene encodes a member of the EF-hand calcium-binding protein family. It is one of three genes which encode an identical calcium binding protein which is one of the four subunits of phosphorylase kinase. Two pseudogenes have been identified on chromosome 7 and X. Multiple transcript variants encoding different isoforms have been found for this gene.[provided by RefSeq, Oct 2009]