We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
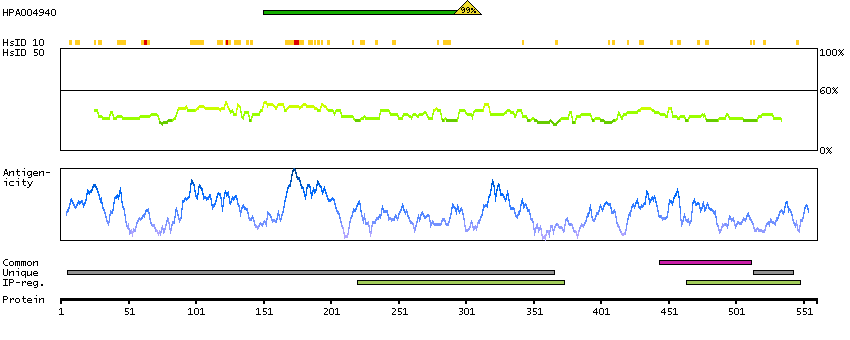
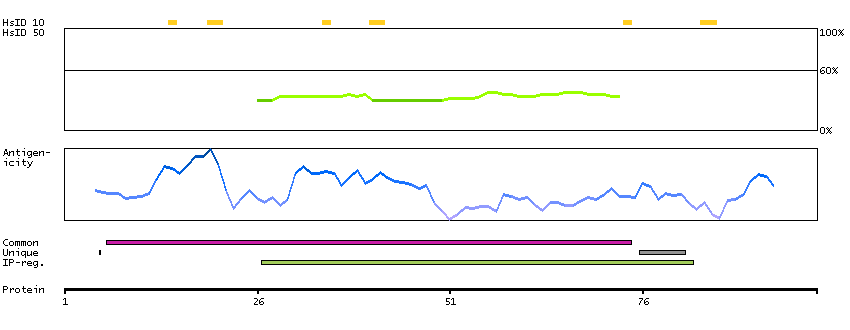
The protein encoded by this gene was identified on the basis of its interaction with MAP3K12/DLK, a protein kinase known to be involved in the induction of cell apoptosis. This gene product contains a leucine zipper, which is a characteristic motif of transcription factors, and was shown to exhibit strong transactivation activity when fused to Gal4 DNA binding domain. Overexpression of this gene interfered with MAP3K12 induced apoptosis. [provided by RefSeq, Jul 2008]