We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
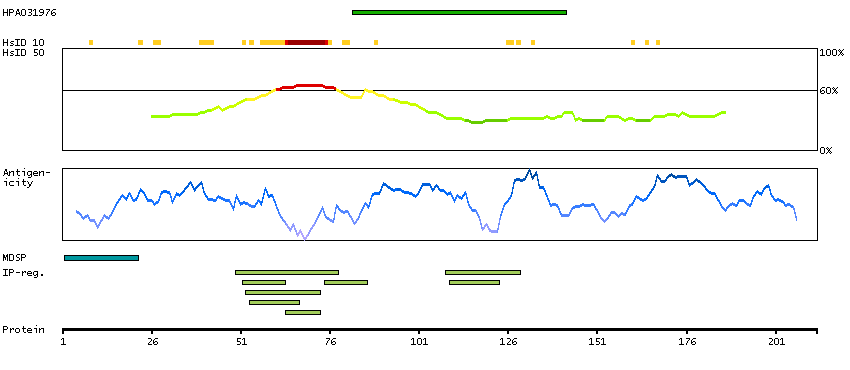
This gene encodes a preproprotein that is proteolytically processed to generate a secreted peptide that belongs to the endothelin/sarafotoxin family. This peptide is a potent vasoconstrictor and its cognate receptors are therapeutic targets in the treatment of pulmonary arterial hypertension. Aberrant expression of this gene may promote tumorigenesis. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Oct 2015]